

FDA's HSP/BIMO Initiative Accomplishments - Update (2009)
March 25, 2009
The Food and Drug Administration (FDA) is providing a progress report on the agency's Human Subject Protection (HSP)/Bioresearch Monitoring (BIMO) Initiative.1 Launched in 2006 as a part of the Critical Path Initiative,2 the HSP/BIMO Initiative is aimed at modernizing and strengthening the agency's oversight and protection of subjects in clinical trials and the integrity of resulting data. The HSP/BIMO Initiative encompasses all FDA-regulated clinical trials, that is, those related to human drugs and biological drug products, devices, foods, and veterinary medicine. The overarching goals of the agency's BIMO program are to protect the rights, safety, and welfare of subjects involved in FDA-regulated clinical trials; to determine the accuracy and reliability of clinical trial data submitted to FDA in support of research or marketing applications; and to assess compliance with FDA's regulations governing the conduct of clinical trials, including those for informed consent and ethical review.
After performing an inventory of its programs, FDA engaged the participation of internal and external stakeholders, conducted workshops, and created other opportunities for public input. During the past two years, the agency has been working diligently to develop and issue new regulations and guidance to further improve the conduct of clinical trials and enhance the protection of participating human subjects. Some of these documents are addressed to study sponsors, clinical investigators, and institutional review boards; others focus on FDA's internal procedures and oversight activities. The agency has also revamped its processes for taking regulatory actions against clinical investigators who repeatedly or deliberately fail to comply with FDA's regulations governing the conduct of clinical trials. Discussed below are the major accomplishments to date.
Highlights:
New Regulations
- Registration of Institutional Review Boards (IRBs), 21 CFR 56.106.3 This new regulation, which requires IRBs to register through a system maintained by the Department of Health and Human Services (HHS), will make it easier for FDA to identify IRBs that review FDA-regulated research and convey educational information to them. (Issued January 2009; All IRBs will need to comply with the initial registration requirement and, if necessary, make required revisions to their registrations by September 14, 2009.)
- Requirements for Acceptance of Data from Foreign Clinical Trials Not Conducted under IND, 21 CFR 312.120.4 This final rule updates the standards for FDA to accept, as support for an investigational new drug application (IND) or application for marketing approval, a well-designed, well-conducted, non-IND foreign clinical study if it was conducted in accordance with Good Clinical Practice (GCP), including review and approval by an independent ethics committee (IEC), and provided that FDA is able to validate the study data through an onsite inspection, if necessary. The final rule's emphasis on GCP helps to ensure effective human subject protection, provides greater assurance of the quality and integrity of data obtained from these studies, and reflects progress in, as well as the success of, FDA's efforts to harmonize international standards for the conduct of clinical research. (Issued April 2008; effective October 2008)
New Guidance
- Final Guidance, "Adverse Event Reporting to IRBs -- Improving Human Subject Protection."5 This guidance is intended to alleviate the burden IRBs face from multiple individual adverse event reports submitted without analysis or context as to their relevance to subject safety. The guidance provides recommendations to IRBs, sponsors, and investigators on improving the usefulness of the adverse event information submitted to IRBs. (January 2009)
- Final Guidance, "Data Retention When Subjects Withdraw from FDA-Regulated Clinical Trials."6 This guidance explains the basis for FDA's policy that already accrued data, related to individuals who cease participating in a study, are to be maintained in the study database. Maintenance of this data helps to ensure that the studies upon which FDA relies for its regulatory decisions are scientifically valid and ethical. (December 2008)
- Draft Guidance, “Humanitarian Device Exemption (HDE) Regulation: Questions and Answers.”7 The guidance addresses a number of topics related to the submission and review of HDE applications, including IRB review of HDEs and the new pediatric provisions of the Pediatric Medical Device Safety and Improvement Act of 2007. (August 2008)
- Draft Guidance, "Frequently Asked Questions - Statement of Investigator (Form FDA 1572)."8 This document addresses questions related to completion of the form and its applicability to studies involving non-U.S. sites. (July 2008)
- Final Guidance, “Computerized Systems Used in Clinical Trials.”9 This guidance provides recommendations regarding the use of computerized systems in clinical investigations. It applies to records in electronic form that are used to create, modify, maintain, archive, retrieve, or transmit clinical data required to be maintained or submitted to FDA. The goal is to assist industry in ensuring confidence in the reliability, quality, and integrity of electronic source data and source documentation (i.e., electronic records). (May 2007)
Improvements to FDA's Internal Procedures
FDA established an internal task force to ensure that all pending and future recommendations related to the agency's oversight of clinical trials raised by Congress, the HHS Office of the Inspector General (OIG)10 and the General Accounting Office (GAO) are fully addressed. As recommended, FDA established procedures to enhance communication between its field and headquarters staff, developed criteria for initiating certain regulatory actions, and provided additional training to its staff in a number of areas. Specifically, FDA:
- Updated the Compliance Program Guidance Manual (CPGM) chapter on Clinical Investigator Inspections (7348.811)11 to improve communication among FDA staff before, during, and after an inspection and to more clearly define the threshold criteria for issuing Warning Letters or notices initiating disqualification proceedings to clinical investigators. Revisions also seek to ensure that clinical investigators are providing required financial disclosure information to trial sponsors. (December 2008)
- Developed and implemented internal procedures and guidance documents to provide direction to inspectional staff and ensure consistency and transparency related to FDA's process for disqualifying clinical investigators who repeatedly or deliberately fail to comply with regulations for human subject protection and the conduct of clinical investigations. This includes:
- Revising the Regulatory Procedures Manual (RPM)12 to include a chapter on disqualification of clinical investigators that describes the procedures to be followed from the conclusion of an inspection through the issuance of a Notice of Opportunity for Hearing (NOOH). The chapter provides clear timeframes for all parties involved in the process to allow equitable, timely, and consistent processing of disqualification matters. (January 2009)
- Revision of FDA's Staff Manual Guide (SMG) to provide a new chapter on disqualification matters involving clinical investigators (SMG 7711).13 The SMG describes the procedures and timeframes for conducting a regulatory hearing to determine if the investigator should remain eligible to receive investigational products. (July 2008)
- Convened a working group to discuss ways to improve the agency's debarment process. Under section 306 of the Federal Food, Drug, and Cosmetic Act, FDA has the authority (among other things) to debar certain persons from the drug industry, such as companies and individuals convicted of crimes related to the drug approval process. Debarment is thus integral to the agency's clinical trial oversight activities.
- In the past, the agency's exercise of its debarment authority had not been centralized. FDA, through the efforts of the working group, has now finalized an SMG that consolidates the authority to initiate and pursue debarment actions in the Office of Enforcement, within the agency's Office of Regulatory Affairs. This consolidation will promote efficiency and consistency in the debarment process.
- The new SMG also establishes specific procedures and timeframes for initiating, pursuing, and finalizing debarment actions. These new procedures will further ensure a timely and consistent debarment process.
While FDA has already implemented a number of changes to its clinical trial oversight activities, the agency continues to look for new approaches to these responsibilities. The agency has been striving to implement a quality systems approach that will provide for continuous process improvements, thus building quality into the clinical trial upfront rather than assessing it at trial completion. To further this effort, FDA co-sponsored two public meetings where in-depth discussions of quality systems in clinical trials were held.
Clinical Trial Transformation Initiative (CTTI)14
Recognizing the need to modernize the clinical trials enterprise, FDA and Duke University formed a public-private partnership that aims to identify practices that through broad adoption will increase the quality and efficiency of clinical trials. CTTI includes representation from government, industry, patient advocacy groups, professional societies, and academia. Current projects include the assessment of various clinical trial monitoring methods to assist sponsors in selecting the most appropriate techniques for a specific trial and development of “best practices” for analyzing and reporting results of clinical trials.
International harmonization, capacity-building, and outreach activities
- FDA staff participated both as faculty and students at a training program for inspectors of the European Medicines Agency (EMEA). The agency is also exploring ways to improve communications with the EMEA, including information sharing agreements and personnel exchanges to gain a better understanding of each organization's regulatory processes and activities.
- FDA staff serve as the U.S. representative to international working groups on GCP.15 The agency actively participates in training international health authorities related to inspection of clinical trials, human subject protection, GCP, and international standards for data quality, thus leveraging resources by building a local framework for GCP regulation and oversight in other regions.16
Activities in Progress
Guidance in Development
- Final Guidance, "Investigator Responsibilities - Protecting the Rights, Safety, and Welfare of Study Subjects."17 FDA is developing this guidance to emphasize investigator's responsibilities when conducting FDA-regulated clinical trials and help them better meet these responsibilities, including delegating study tasks only to appropriately qualified personnel.
- Final Guidance, "Frequently Asked Questions - Statement of Investigator (Form FDA 1572)."18 FDA is reviewing comments submitted to the docket and will revise the draft guidance as needed.
- Final Guidance, “Information Sheet Guidance on IRB Continuing Review after Clinical Investigation Approval.”19 This document will describe in more detail, the criteria, process, and frequency of continuing review to help assure the protection of rights and welfare of study subjects.
- Final Guidance, "Exception from Informed Consent Requirements for Emergency Research."20 FDA is reviewing comments submitted to the docket and presented at an open public meeting on the draft guidance in order to finalize the document.
- Draft Guidance, “Information Sheet Guidance for IRBs, Clinical Investigators, and Sponsors; A Guide to Informed Consent.” This document will describe in detail basic and additional elements of informed consent and will include topics such as review of patient records, children as subjects, and subject participation in more than one study to help assure the protection of rights and welfare of study subjects.
Modernizing our Regulations
At the direction of the HSP/BIMO Council, FDA convened a working group to discuss the need for updating the agency's Good Laboratory Practice for Nonclinical Laboratory Studies regulation (21 CFR Part 58) to better reflect current practices. This agency-wide working group, which includes representatives from the Center BIMO programs, the National Center for Toxicological Research, ORA, and the Office of the Commissioner, has conducted a thorough review of the regulation, discussed stakeholder input, and will soon present its suggestions for modernizing the regulation to the HSP/BIMO Council.
BIMO Inspections for Fiscal Year 2008
Each year, FDA's field staff conduct on-site inspections of BIMO facilities, including sponsors, monitors, clinical investigators, IRBs, and laboratories that conduct nonclinical safety studies (including animal toxicity studies) to support FDA-regulated research. The agency performs these inspections to evaluate the inspected party's practices and procedures to determine compliance with applicable regulations. FDA has generally conducted the majority of its BIMO inspections in association with the submission of a marketing application or as a part of its investigation of a complaint. Recently, the agency has been shifting more of its resources to inspections of ongoing studies. This will allow the agency to identify potential problems while the study is still going on, thus enabling the implementation of corrective actions to minimize risks to human subjects and preserve the integrity of the clinical trial. FDA has also been improving its follow-up of violative inspections and working to identify alternative methods to select clinical investigator sites for inspection, such as risk-based approaches and using statistical evaluations to identify sites of interest.
Summary information about FDA's domestic inspectional activities for fiscal year 2008 is presented in the following:.

Figure 1
Figure 1 depicts the total number of inspections that were completed by the Office of Regulatory Affairs' field investigators as well as the numbers completed for each of the assigning Centers. (BEQ denotes bioequivalence studies.)
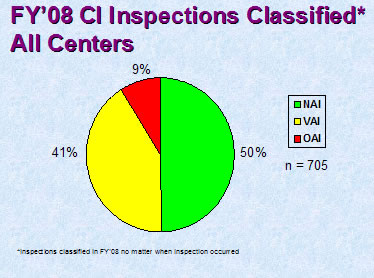
Figure 2
Figure 2 depicts the compliance classification of Establishment Inspection Reports (EIRs) for inspections of clinical investigators that were classified by the assigning Center in FY 2008. (NAI denotes No Action Indicated, VAI denotes Voluntary Action Indicated, and OAI denotes Official Action Indicated.)

Figure 3
Figure 3 depicts the compliance classification of EIRs for inspections of IRBs that were classified by the assigning Center in FY 2008.

Figure 4
Figure 4 depicts the compliance classification of EIRs for inspections of sponsors that were classified by the assigning Center in FY 2008.
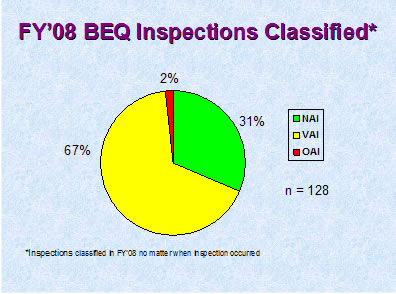
Figure 5
Figure 5 depicts the compliance classification of EIRs for inspections of bioequivalence studies that were classified in FY 2008.
Footnotes
1 The HSP/BIMO Council, which is overseeing this initiative, includes representatives from each of FDA's Centers, the Office of Regulatory Affairs (ORA), and the Office of the Commissioner (OC).
2 FDA Announces New Initiative to Modernize the Regulation of Clinical Trials and Bioresearch Monitoring
3 Institutional Review Boards; Registration Requirements
5 Adverse Event Reporting to IRBs Improving Human Subject Protection
6 Data Retention When Subjects Withdraw from FDA-Regulated Clinical Trials
7 Draft Guidance for HDE Holders
8 Frequently Asked Questions - Statement of Investigator Form FDA 1572
9 Computerized Systems Used in Clinical Investigations
10 Examples of recent OIG reports include FDA Oversight of Clinical Investigators (June 2000; www.oig.hhs.gov/oei/reports/oei-05-99-00350.pdf ); The Food and Drug Administration's Oversight of Clinical Trials (September 2007, www.oig.hhs.gov/oei/reports/oei-01-06-00160.pdf ); FDA's Oversight of Clinical Investigators' Financial Information (January 2009, www.oig.hhs.gov/oei/reports/oei-05-07-00730.pdf ).
11 Compliance Program Guidance Manual For FDA Staff
12 Regulatory Procedures Manual
13 SMG 7711
14 www.trialstransformation.org
15 For example, Pan American Health Organization's GCP Working Group.
16 Examples include Asia Pacific Economic Cooperation, Bangkok, May 2008; India, September 2008 and May-June 2009; Multinational Clinical Trial Symposium for Middle East/North African States, Jordan, October 2008.
17 Draft guidance is available at: Protecting the Rights, Safety, and Welfare of Study Subjects - Supervisory Responsibilities of Investigators
18 Draft guidance is available at: Exception from Informed Consent Requirements for Emergency Research. The link to the draft guidance has been updated to link instead to the final guidance issued in April 2013.
19 Draft guidance is available at: Continuing Review After Study Approval
20 Draft guidance is available at: Exception from Informed Consent Requirements for Emergency Research. The link to the draft guidance has been updated to link instead to the final guidance issued in April 2013.