

GUIDANCE DOCUMENT
Medical Device Classification Product Codes - Guidance for Industry and Food and Drug Administration Staff April 2013
- Docket Number:
- FDA-2011-D-0916
- Issued by:
-
Guidance Issuing OfficeCenter for Biologics Evaluation and ResearchCenter for Devices and Radiological Health
Document issued on: April 11, 2013
The draft of this document was issued on January 3, 2012.
For questions about this document regarding CDRH or CBER-regulated devices, contact the CDRH 510(k) Program at 510k_program@fda.hhs.gov or (301) 796-5640.

U.S. Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Center for Biologics Evaluation and Research
Preface
Public Comment
You may submit written comments and suggestions at any time for Agency consideration to the Division of Dockets Management, Food and Drug Administration, 5630 Fishers Lane, rm. 1061, (HFA-305), Rockville, MD, 20852. Submit electronic comments to http://www.regulations.gov. Identify all comments with the docket number listed in the notice of availability that publishes in the Federal Register. Comments may not be acted upon by the Agency until the document is next revised or updated.
Additional Copies
Additional copies are available from the Internet. You may also send an e-mail request to CDRH-Guidance@fda.hhs.gov to receive a copy of the guidance. Please use the document number 1774 to identify the guidance you are requesting.
Additional copies of this guidance document are also available from the Center for Biologics Evaluation and Research (CBER) by written request, Office of Communication, Outreach and Development (HFM-40), 1401 Rockville Pike, Suite 200N, Rockville, MD 20852-1448, by telephone, 1-800-835-4709 or 301-827-1800, by email, ocod@fda.hhs.gov, or from the Internet at http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/default.htm.
Table of Contents
- Introduction
- Definition
- Use of Classification Product Codes in Premarket Review
- Use of Classification Product Codes in Post Market Review
- Product Codes for Licensed Devices in CBER
- Appendix A. Frequently Asked Questions
- Appendix B. Resources
Medical Device Classification Product Codes - Guidance for Industry and Food and Drug Administration Staff
This guidance represents the Food and Drug Administration's (FDA's) current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations. If you want to discuss an alternative approach, contact the FDA staff responsible for implementing this guidance. If you cannot identify the appropriate FDA staff, call the appropriate number listed on the title page of this guidance.
Introduction
Since the passage of the May 28, 1976 Medical Device Amendments to the Food, Drug and Cosmetic Act (FD&C Act), the Classification Regulation Panels (21 CFR Parts 862-892) have been the basis for the Center for Devices and Radiological Health’s (CDRH’s) Classification Product Code structure and organization. In order to respond to the evolution of device technology, classification product codes were created to assist in accurate identification and tracking of current medical devices and to allow for tracking of and easy reference to predicate device types. Classification product codes are used by FDA to obtain quality and reliable data, and perform analyses that are often reported to Congress, the Government Accountability Office (GAO), the general public, the media, and industry. Classification product codes are also used throughout the total product life cycle (TPLC) as they connect all medical device databases.
This document describes how device product codes are used in a variety of FDA program areas to regulate and track medical devices regulated by the Center for Devices and Radiological Health (CDRH) and the Center for Biologics Evaluation and Research (CBER). This document is limited to medical devices as defined in section 201(h) of the Federal Food Drug & Cosmetic (FD&C) Act and does not discuss classification product codes used to regulate non-medical electronic radiation emitting products.
The scope of this document includes devices described in the existing classifications under 21 CFR Parts 862-892. It also describes how the product code builder developed by FDA’s Office of Regulatory Affairs is used for devices that are licensed under the Public Health Service Act (PHS Act), and currently do not have product codes generated under classification regulation panels. It also covers unclassified devices and devices not yet classified.1
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
Definition
Classification product codes are a method of internally classifying and tracking medical devices. CDRH and a subset of CBER regulated medical device product codes consist of a 3 letter combination which associates a device’s type with a product classification designated for the application. Classification product codes and information associated with these devices, such as names and attributes, are assigned by CDRH to support their regulation.
Use of Classification Product Codes in Premarket Review
1. General Uses of Classification Product Codes for Premarket Submissions
- Classification product codes help to delineate technology and indication subgroups within a regulation. They can also serve to categorize unclassified or Class III (PMA) devices. Some of these subgroups may require different levels of evidence to support marketing clearance or approval, or specific warnings in the labeling. Classification product codes are also useful to identify devices for subsequent changes such as reclassification. If a device is reclassified (e.g., from Class III to Class II) the classification product code may stay the same or a new classification product code may be created.
- Classification product codes are used for internal tracking purposes, such as adverse event monitoring or compliance actions. Product codes may be created and assigned for these purposes.
- Classification product codes are assigned and maintained by the Agency. The submitter of the premarket submission selects a product code based on the identified predicate device(s). The proposed product code is reviewed by FDA staff for accuracy. If the proposed product code is incorrect, or a more appropriate product code should be used, the reviewer will change the product code and notify the applicant. In the case of adverse event reporting, when an incorrect product code is used or a new tracking product code is created, the Agency will assign the most accurate product code to the adverse event report.
- Proposed classification product codes cited in premarket submissions are used in the initial assignment to the appropriate review branch/division. They are also used to determine the review panel for the device.
- As new classification product codes are created, a device may be re-assigned into a new product code. If this occurs, FDA will send a corrected substantially equivalent or approval letter to the manufacturer of the marketed device to notify them of the new classification product code.
- Guidance documents often define their scope by referencing the classification product codes to which they apply.
2. Premarket Notification [510(k)] Devices
- Classification – Product codes are assigned within established classification regulations as described in 21 CFR Part 860. They are also assigned to unclassified devices and not-classified devices. An unclassified device is a pre-amendments device for which a classification regulation has not been promulgated. Until the unclassified device type has been formally classified and a regulation established, marketing of new devices within this type will require submission of a 510(k) premarket notification to CDRH or CBER. Once classified, these devices may require submission of a PMA, a 510(k), or be exempt from any premarket submission. A not-classified device is a post-amendments device for which the Agency has not yet reviewed a marketing application or for which the Agency has not made a final decision on such a marketing application.
- Predicate Devices –To demonstrate substantial equivalence and therefore obtain clearance of a 510(k) submission, a comparison to a predicate device is provided. Selecting a predicate device with the same classification product code as the proposed device is usually most appropriate.
- Assignment – The reviewer will assign a classification product code based on the regulation (if relevant) or the device intended use, indications for use or technology. The most common method of assignment is to use an existing product code from the product code database. A device will be assigned an existing classification product code when it has the same intended use, indications for use, and relies on technology that does not raise new safety and effectiveness questions. However, if the proposed device differs significantly from the predicate device with respect to technology, intended use or indications for use or is found not substantially equivalent (NSE), a new product code should be assigned. The 510(k) summary will include all classification product codes considered relevant. The 510(k) clearance letter will specify one primary product code for the device and may include subsequent product codes that address additional features or functions of the device. The primary classification product code should correspond with the regulation and class applicable to the device and should be used in all postmarket correspondence as needed. The accurate use of this product code in all actions including premarket submissions, adverse event reporting and compliance actions is important to ensure accurate communication with the Agency and to avoid potential negative impacts such as delays in delivery or shipment. Multiple subsequent product codes can be used even if they fall under a different regulation and class. If multiple product codes are assigned to a device, the product code that corresponds with the highest regulatory class or, if the regulatory class is the same, the product code corresponding to the most relevant technology will be used as the primary product code.
- Evolution – As technology changes and 510(k) review practice evolves, some classification product codes may become obsolete. In some cases, product code definitions may be updated to accommodate new technology. Obsolete product codes will still be maintained in the public database for tracking purposes. Predicates with obsolete product codes will remain under the same regulation and may still be used as valid predicates. A corrected clearance letter should be issued when a device with an obsolete product code is assigned a new product code. New classification product codes should be created for a 510(k) when a device has a new intended use or when a device incorporates technology that raises new questions of safety and effectiveness. In the case where a device is found not substantially equivalent (NSE) to a predicate device; the classification product code describing the device will be placed in the product code database for future reference. Additionally, new product codes may be created for tracking purposes for a specific technology or device area. Please see the FAQ section of the guidance for instruction on where to seek clarification on an assigned product code.
- Other – Classification Product codes can be used to help classify other premarket submission variations.
- Third party eligible 510(k) submissions – The classification database can be searched by product code to determine which devices may be eligible for third party review. For more information on the classification database, please see the “Resources” section of this guidance document.
- Evaluation of Automatic Class III Designation (De Novo) (513(f)) requests)
– When a De Novo petition is granted and the device is reclassified, the product code assigned when the 510(k) was submitted and determined to be NSE will be used.2 Note that Section 607 of the Food and Drug Administration Safety and Innovation Act (FDASIA) amended Section 513(f)(2) of the FD&C Act to allow submission of a De Novo petition without submission of a 510(k) and subsequent NSE if there is no legally marketed device upon which to base an SE determination. In such cases, a new product code will be assigned at the time that the De Novo petition is granted. - Kits – Product codes have been assigned to established convenience kits that are outlined in FDA’s Convenience Kits Interim Regulatory Guidance. The list of established convenience kits can be found in the guidance.
3. Premarket Approval (PMA) Devices and Humanitarian Device Exemption (HDE) Devices
- The use of product codes for PMA and HDE devices is similar to their use for 510(k) devices. A product code is assigned to the device upon approval and is included in the subject of the approval letter of a PMA or HDE.
- Class III devices are not always classified under a specific regulation, but may have a product code assigned. When a regulation for the device type exists, Class III devices are assigned under a specific regulation, like Class I and II devices. When Class III devices are reclassified to Class II devices, a regulation is created and the product code may remain the same or a new product code may be created.
- Modifications to an existing Class III device that result in the submission of a PMA supplement, such as a change in indications for use, which would be submitted through a panel track PMA supplement, may require creation and assignment of a new product code for the modified device.
- The appropriate product code should be cited in submissions of annual reports.
- For HDE submissions, the eligibility for humanitarian exemption is not based on the product code of the proposed device. The proposed device, along with the indications for use, must be designated as a Humanitarian Use Device by the Office of Orphan Products before an HDE can be submitted to CDRH or CBER.3
4. Investigational Device Exemption (IDE)
- A classification product code may be assigned to a device that is the subject of an IDE submission and included in the approval letter for an IDE if the device falls in a known device area with established technology or will be tracked internally for a specific technology, patient population or device area.
- The classification product code assigned to a device that is the subject of an IDE application is primarily used for internal tracking purposes and may differ from that of the final PMA approval or 510(k) clearance. In many instances, a product code will not be assigned to an IDE submission.
- For devices where an established classification product code is known, the proposed product code should be specified in the submission. For novel devices, a new classification product code will generally be assigned by FDA at the time of PMA approval/510(k) clearance.
5. Request for Classification (513(g)) Applications
Section 513(g) of the FD&C Act (21 U.S.C. 360c(g)) provides a means to obtain the Agency's views about the classification and the regulatory requirements that may be applicable to a particular device. FDA’s response to a 513(g) request will include, in part, the Agency's assessment, based on the information submitted in the request, as to the generic type of device (e.g., classification regulation) that the requester's device appears to be within (if any); the class of devices within that generic type; and whether a PMA, 510(k), or neither is required in order to market devices of the particular class within that generic type. However, a classification product code is not generally specified in a 513(g) classification for devices that will require a premarket submission. The recommendation in response to the 513(g) submission only lists proposed devices within a given regulation. Devices that are Class I exempt or Class II exempt may receive a classification product code as part of this recommendation.
Use of Classification Product Codes in Post Market Review
1. Adverse Events
Classification product codes are a key element in the reporting of adverse events and product problems in medical device reports (MDRs). Though not clearly requested in the 3500A mandatory reporting form (MedWatch Form), it is common practice for the reporter to indicate the product code along with the common name of the device in section D2 of the 3500A form. The addition of the product code by mandatory reporters supports 21 CFR 803.52(c)(2) by describing the type of product. The classification product code used by reporters should be the primary product code associated with the device for which the report is being made. To improve the quality of MDRs, we recommend that reporters include the premarket submission number (if applicable) in section G5 of the 3500A form to further link the device to its original classification.
In cases where the classification product code is not known by the reporter and an MDR is submitted without one, CDRH assigns the appropriate product code to the MDR based on the brand name (section D1), common device name (section D2), or premarket submission number (section D5). However, it is preferred that the reporter of the MDR provide the product code with which CDRH classified the device (in the case of Class II and III devices).
2. Import/Export
A. Office of Regulatory Affairs (ORA) Product Code Builder
In order to ensure that a medical device is in compliance with FDA regulatory requirements, importers/brokers/filers are required to submit certain import information. One data element that is required to be provided is the product code. If the product code is unknown, importers/brokers/filers can use the Office of Regulatory Affairs’ (ORA) Product Code Builder to formulate a product code for the product they are importing. In addition, the CDRH Product Classification Database can be used to look up a device's definition and regulatory requirements, neither of which is provided in the ORA Product Code Builder. As new product codes are created by CDRH and old ones modified, ORA’s Division of Compliance Systems (DCS) is notified, and the Product Code Builder is updated. The product code used for the FDA import admissibility review process is formatted differently than the classification product code used by CDRH. There is no definitive meaning for the three digit classification product codes in CDRH’s Product Classification Database. However, ORA’s Product Code Builder uses a seven digit product code, rather than the three letter combination found in the product code database. The seven digit product codes encompass devices, foods, drugs, biologics, and cosmetics and each digit signifies a particular description. For example, FRN is the product code assigned to Pump, Infusion in CDRH’s Product Classification Database. The same product code translates to 80F--RN in ORA’s Product Code Builder. The two numbers at the beginning of the seven-digit product code represent the medical specialty panel classified for the device.
B. Import Entry Process
The classification product code helps the FDA import entry reviewer determine what information he/she should verify to ensure the medical device meets all FDA regulatory requirements (e.g., registration, listing, clearance/approval numbers).
Classification product codes are also used by FDA to designate products for Import Alerts. Import Alerts identify problem commodities, shippers and importers, and provide guidance for import coverage.
3. Recalls
Classification product codes are an important aspect in reporting recalls, corrections and removals. They are used to ensure correct device identification to determine which group of the FDA will be responsible for review and classification of the recall, correction or removal. Though not clearly required in 21 CFR 806.10, the addition of the product code by device manufacturers and importers supports the requirement in 21 CFR 806.10(c)(4) to provide the device’s marketing status, since the product code is assigned during pre-market review or approval (510(k), PMA, HDE or EUA (Emergency Use Authorization)) and indicates the regulatory classification of the device. The device manufacturers and importers should provide the primary product code associated with the recalled device, unless the secondary product code is more specific. In cases where the product code is not known by the device manufacturer or importer, the product code assigned to the device during device listing (Establishment Registration and Device Listing) should be used.
4. Establishment Registration and Device Listing
A. Determining How FDA Will Classify Your Device
If your product is considered a medical device, you must determine how your device is classified by FDA for the purposes of registration and listing.4 Step- by-step instructions on determining how FDA will classify your device can be found on the FDA website.5 Here you will identify the correct device name, regulation number, and classification product code for your device. You will use this information to list your device in the FDA Unified Registration and Listing System (FURLS)/Device Registration and Listing Module (DRLM). If you need assistance with determining if your product is a device or the appropriate classification for the device, please contact the Division of Small Manufacturers, International, and Consumer Assistance (DSMICA) by email at dsmica@fda.hhs.gov to receive assistance.
B. Listing Your Device
Instructions for listing your device are posted on our website6 under the “Initial Registration” heading.
i. Exempt Devices
Devices that are notsubject to premarket notification (510(k)), premarket approval (PMA) or HDE requirements are considered exempt devices. You will need to determine the classification product code for your exempt device before you can list the device in FURLS/DRLM. You can identify the product code by searching the Product Classification database. Once you know the classification product code, you can list the device.
ii. Non-Exempt Devices
Devices that are subject to premarket notification or premarket approval requirements [510(k), PMA] are considered non-exempt devices. If your device requires premarket notification clearance or approval (510(k) or PMA), please remember the following:
- You cannot list the device until the 510(k) or PMA has been cleared or approved.
- You need your premarket submission number to list your device in FURLS/DRLM.
- You should list your device with the Premarket Submission Number (510(k), PMA). The classification product code that was assigned on your clearance/approval letter will appear on your listing.
- If you believe the classification product code in the CDRH Corporate database is not correct, then you will need to contact the Program Operations Staff (POS) in ODE for assistance with correcting this information. Contact information is listed in the Resource Section of this guidance document.
iii. Devices Licensed as Biological Products under the PHS Act
Manufacturers of devices regulated by CBER under the PHS Act, i.e., manufacturers of licensed in vitro diagnostics including donor screening tests, should follow the registration and listing requirements in 21 CFR 607.20 and refer to the section “Product Codes for Licensed Devices in CBER” later in this guidance.
C. Enforcement Discretion
Devices for which FDA applies enforcement discretion and therefore pre-market review is not necessary, require additional instructions before they can be listed in FURLS/DRLM. You should contact the Registration and Listing Staff by email at reglist@cdrh.fda.gov to obtain the additional instructions prior to attempting to list such a device. A few devices for which FDA applies enforcement direction are certain kits and export only devices.
i. Convenience Kits
You should consider the following before listing a convenience kit:
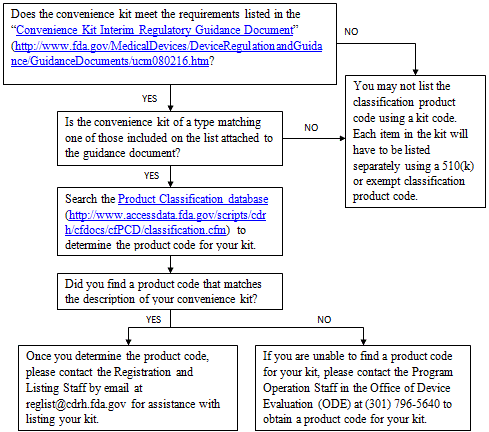
ii. US Manufacturers of Export Only Devices
US manufacturers of Export Only Devices are required to list the devices that they export to a foreign country.7 However, 510(k) clearance or PMA/HDE approval is not needed for the device to be exported to the foreign country. Some manufacturers of export only devices may market the same device in the United States. In this case, the manufacturer uses the classification product code assigned to the cleared/approved device to list their device in FURLS/DRLM. If the manufacturer is not marketing the same device in the United States, they should contact the Program Operations Staff and obtain a new classification product code for “export only”. Once a new classification product code is assigned, please contact the Registration and Listing Staff by email at reglist@cdrh.fda.gov for assistance with listing devices that are being exported to a foreign country.
Product Codes for Licensed Devices in CBER
CBER regulates a range of devices, most of which are subject to the FD&C Act only. However, some CBER devices are licensed under the PHS Act (e.g., in vitro diagnostic tests required for blood donor screening and related blood banking practices). Consequently, licensed devices are subject to the regulations outlined in 21 CFR Part 600 in addition to those in 21 CFR Part 800. The different regulatory requirements impact classification product codes as well.
In contrast, product codes for licensed devices are generated using the principles which apply to most other FDA regulated products (please refer to the Import section). As is the case with other biologics, licensed devices are classified under industry code 57. For example, a licensed blood donor screening assay for Hepatitis B surface antigen has a six character product code of the following structure: 57 V H-05. Of note, the letter in the subclass element can either be an H, I or L, depending whether this is a final product, intended for further manufacture or product sample for testing/lot release.
Appendix A. Frequently Asked Questions
1. I have a device for export only. I cannot find the appropriate classification product code in the classification database, how would I be able to successfully list the device?
Answer: You should contact the Program Operations Staff in order to have a new product code created for the intent of export only.
2. How do I search for a classification product code?
Answer: You can search for a product code using the Product Classification Database on FDA’s website: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/PCDSimpleSearch.cfm
3. I have searched the product code database and do not find a suitable classification product code for my device. What should I do?
Answer: You can contact the appropriate review division within CDRH/CBER. You may also submit a Request for Classification (513(g)) application.8
4. How do I update/change a classification product code?
Answer: Contact the Product Code Coordinator at 301-796-5640.
5. What do I do if the classification product code on my 510(k) substantially equivalent (SE) or PMA approval letter is incorrect?
Answer: Contact the appropriate review division within CDRH/CBER. The contact information will be at the bottom of the SE letter or approval letter. If the classification product code is incorrect, they will make the correction and send you a corrected clearance or approval letter.
6. What happens to the classification product code when the device is reclassified?
Answer: The product code database will be updated to reflect the new information and the affected firms will be notified in writing in a timely manner.
7. What should I do if I notice an error in the product classification database?
Answer: Contact the product code coordinator at 301-796-5640 and the database will be corrected. The product classification database found at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/PCDSimpleSearch.cfm is updated weekly.
8. What is the difference in review time if a submission requires the creation of a new product code?
Answer: There is no difference in the review time when a new product code is needed.
9. Does FDA work with the Centers for Medicare and Medicaid Services (CMS) on product codes for reimbursement issues?
Answer: No. FDA will provide information to CMS regarding the regulatory requirements associated with a specific product code, but is not further involved in the reimbursement process. For reimbursement issues, CMS should be contacted directly.9
10. Is it helpful to include a classification product code on my adverse event report?
Answer: Absolutely, it improves the quality of data reporting and appropriate routing of the report for analysis.
Appendix B. Resources
- Program Operations Staff/Office of Device Evaluation - 301-796-5640
- Product Classification Database
- Office of Regulatory Affairs (ORA) Product Code Builder
- Device Advice – Device Regulation and Guidance
- Device Classification
- Medical Device Listing
- Convenience Kits Guidance
- Medical Device Reporting (MDR)
- Industry Procedures for Section 513(g) Requests for Information
- Evaluation of Automatic Class III Designation, Guidance for Industry and CDRH Staff (De Novo)
1 An unclassified device is a pre-amendments device for which a classification regulation has not been promulgated. Unclassified devices require submission of a 510(k) premarket notification to CDRH. A not-classified device is a post-amendments device for which the Agency has not yet reviewed a marketing application or for which the Agency has not made a final decision on such a marketing application. A pre-amendments device is a device that was on the market prior to the enactment of the Medical Device Amendments to the FD&C Act on May 28, 1976.
2 See Guidance for Evaluation of Automatic Class III Designation.
3 For additional information regarding HUD designation, refer to information on the FDA website.
4 See 21 CFR Part 807.
5 Device Advice: Comprehensive Regulatory Assistance
7 See 21 CFR Part 807.
8 Refer to the Draft Guidance for Industry and FDA Staff: FDA and Industry Procedures for Section 513(g) Requests for Information under the Federal Food, Drug, and Cosmetic Act on FDA’s website, and the Draft Guidance for Industry and FDA Staff: User Fees for 513(g) Requests for Classification Information.
Submit Comments
You can submit online or written comments on any guidance at any time (see 21 CFR 10.115(g)(5))
If unable to submit comments online, please mail written comments to:
Dockets Management
Food and Drug Administration
5630 Fishers Lane, Rm 1061
Rockville, MD 20852
All written comments should be identified with this document's docket number: FDA-2011-D-0916.