

GUIDANCE DOCUMENT
Changes or Modifications During the Conduct of a Clinical Investigation; Final Guidance for Industry and CDRH Staff May 2001
- Docket Number:
- FDA-2020-D-0957
- Issued by:
-
Guidance Issuing OfficeCenter for Devices and Radiological Health
Document issued on: May 29, 2001

U.S. Department Of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Investigational Device Exemptions Staff
Program Operations Staff
Office of Device Evaluation
Preface
Public Comment
Comments and suggestions may be submitted at any time for agency consideration to Dockets Management Branch, Division of Management Systems and Policy, Office of Human Resources and Management Services, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. When submitting comments, please refer to the exact title of this guidance document. Comments may not be acted upon by the agency until the document is next revised or updated.
For questions regarding the use or interpretation of this guidance contact Sheila Brown at 301-796-6563or e-mail sheila.brown@fda.hhs.gov.
Additional Copies
Additional copies are available from the from the Internet. You may also send an e-mail request to CDRH-Guidance@fda.hhs.gov to receive a copy of the guidance. Please use the document number 1337 to identify the guidance you are requesting.
TABLE OF CONTENTS
- I. Purpose
- II. Background
- III. Changes Requiring Prior Approval vs Changes Subject to 5-Day Notice
- IV. Other Changes to the Investigational Plan
- VI. Conclusion
Attachments
Changes or Modifications During the Conduct of a Clinical Investigation;
Final Guidance for Industry and CDRH Staff
This document is intended to provide guidance. It represents the Agency’s current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind the Food and Drug Administration (FDA) or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute and regulations.
I. PURPOSE
This document provides guidance on the implementation of section 201(a) of the Food and Drug Administration Modernization Act of 1997 (FDAMA) (Pub. L. 105-115), which amended the Federal Food, Drug and Cosmetic Act (the act) by adding section 520(g)(6). This new section of the act establishes criteria that allow sponsors to make certain modifications to the investigational device, including manufacturing changes, and/or to the clinical protocol during the course of the clinical investigation without prior FDA approval.
FDAMA provided for several tools to reduce regulatory burden in bringing new devices to market. Several of these provisions were specifically aimed at the clinical trial process. These include the Agreement/Determination meetings provisions and the least burdensome provisions (sections 201 and 205 of FDAMA, respectively). In addition, new section 520(g)(6) of the act, by permitting sponsors to implement certain changes during the clinical trial without prior approval, may be considered part of Congress’ intent to reduce regulatory burden. Thus, this guidance document is part of the Center’s effort to implement the least burdensome approach in device regulation.
As a result of this new provision, the investigational device exemptions (IDE) regulation (21 CFR 812) was amended. The amendment to the IDE regulation was published in the Federal Register as a proposed rule on July 15, 1998 and as a final rule on November 23, 1998. The preamble to the proposed IDE Modification rule included a detailed discussion of the types of changes that the agency believes are eligible for implementation under this provision as well as the kind of credible information that should be used to support the device and protocol changes. In addition, a discussion of the types of changes for which FDA believes prior approval should be obtained versus those that only require submission in an IDE annual report was also presented.
Although industry has used this new regulatory provision to a certain extent and its use is increasing, it has not been as widely utilized as had been anticipated. Additionally, both industry and CDRH staff have expressed interest in having guidance on the preparation and review of IDE submissions under this regulation. By capturing the discussion provided in the preambles to the proposed and final rules in a guidance document, the agency hopes to encourage IDE sponsors to take advantage of this important new provision.
II. BACKGROUND
Experience has shown that during the course of a clinical investigation, the sponsor of a study will want or need to make modifications to the investigational plan, including the device and/or clinical protocol. These changes may be simple modifications, such as clarifying the instructions for use, or they may be significant changes, such as modifications to the study design or the device materials. Previously, § 812.35(a) Changes in investigational plan of the IDE regulation stated, in part:
A sponsor shall: (1) Submit to FDA a supplemental application if the sponsor or an investigator proposes a change in the investigational plan that may affect its scientific soundness or the rights, safety, or welfare of subjects, and (2) obtain FDA approval under § 812.30(a) of any such change, and IRB approval when the change involves the rights, safety, or welfare of subjects (see §§ 56.110 and 56.111), before implementation.
According to § 812.25 Investigational Plan, the investigational plan includes the purpose of the study, the clinical protocol, a risk analysis, a description of the investigational device, monitoring procedures, labeling, informed consent materials, and institutional review board (IRB) information. Although written guidance on the types of modifications that could have been made without prior FDA approval had not previously been developed, the agency had traditionally permitted changes to all parts of the investigational plan. Thus, the device, the protocol, the monitoring procedures, labeling, etc. could be modified without approval of an IDE supplement if the changes did not affect the scientific soundness of the study or the rights, safety, or welfare of the subjects. According to this past policy, such changes were made by the IDE sponsor and reported in the upcoming annual report.
New section 520(g)(6) of the act, however, required FDA to modify the IDE regulation to explicitly permit certain changes to the investigational device (including manufacturing changes) and to the clinical protocol during the course of the clinical investigation without agency approval of an IDE supplement. Under the new law, the following changes are specifically permitted:
(i) developmental changes in the device (including manufacturing changes) that do not constitute a significant change in design or in the basic principle of operation and that are made in response to information gathered during the course of an investigation; and
(ii) changes or modifications to clinical protocols that do not affect—
(I) the validity of data or information resulting from the completion of an approved protocol, or the relationship of likely patient risk to benefit relied upon to approve a protocol;
(II) the scientific soundness of an investigational plan; or
(III) the rights, safety, or welfare of the human subjects involved in the investigation.
The new law allows these changes to be made if:
(i) the sponsor of the investigation determines, on the basis of credible information (as defined by the agency) that the applicable conditions described above are met; and
(ii) the sponsor submits to the agency, not later than 5 days after making the change or modification, a notice of the change or modification.
Thus, under the new statute, sponsors may modify their investigational device, manufacturing process, and clinical protocol without waiting for agency approval if certain criteria are met. Section 520(g)(6) of the act broadens the criteria beyond that contained in the previous IDE regulation. For device and manufacturing changes, sponsors must ensure that the change does not constitute a significant change in design or principles of operation and that the change is made in response to information gathered during the course of an investigation. For protocol changes, in addition to the previous regulatory requirement that the change not affect the scientific soundness of the study or the rights, safety, or welfare of the subjects, sponsors must now also consider the impact that the change will have on the validity of the data and the risk to benefit relationship. In order for the IDE regulation to reflect the new statutory language and, as required by the new law, FDA modified the IDE regulation. New § 812.35(a) states, in part:
§ 812.35 Supplemental applications. (a) Changes in investigational plan.
(1) Changes requiring prior approval. Except as described in paragraphs (a)(2) through (a)(4) of this section, a sponsor must obtain approval of a supplemental application under § 812.30(a), and IRB approval when appropriate (see §§ 56.110 and 56.111of this chapter), prior to implementing a change to an investigational plan.
(2) Changes effected for emergency use.1 The requirements of paragraph (a)(1) of this section regarding FDA approval of a supplement do not apply in the case of a deviation from the investigational plan to protect the life or physical well-being of a subject in an emergency. Such deviation shall be reported to FDA within 5 working days after the sponsor learns of it (see § 812.150(a)(4)).
(3) Changes effected with notice to FDA within 5 days. A sponsor may make certain changes without prior approval of a supplemental application under paragraph (a)(1) of this section if the sponsor determines that these changes meet the criteria described in paragraphs (a)(3)(i) and (a)(3)(ii) of this section, on the basis of credible information defined in paragraph (a)(3)(iii) of this section, and the sponsor provides notice to FDA within 5 working days of making these changes.
(i) Developmental changes. The requirements in paragraph (a)(1) of this section regarding FDA approval of a supplement do not apply to developmental changes in the device (including manufacturing changes) that do not constitute a significant change in design or in the basic principles of operation and that are made in response to information gathered during the course of an investigation.
(ii) Changes to clinical protocol. The requirements in paragraph (a)(1) of this section regarding FDA approval of a supplement do not apply to changes to clinical protocols that do not affect: (A) The validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the original protocol; (B) The scientific soundness of the investigational plan; or (C) The rights, safety, or welfare of the human subjects involved in the investigation.
(4) Changes submitted in annual report. The requirements of paragraph (a)(1) of this section do not apply to minor changes to the purpose of the study, risk analysis, monitoring procedures, labeling, informed consent materials, and IRB information that do not affect: (i) The validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; (ii) The scientific soundness of the investigational plan; or (iii) The rights, safety, or welfare of the human subjects involved in the investigation. Such changes shall be reported in the annual progress report for the IDE, under § 812.150(b)(5).
1 § 812.35(a)(2) has not been modified. This guidance document does not address deviations from the investigational plan to protect the life or physical well-being of a subject in an emergency (812.35(a)(2)). Such deviations are addressed in the guidance document entitled, "Guidance for the Emergency Use of Unapproved Medical Devices" (50 FR 42866, October 22, 1985) as well as in the guidance entitled, "IDE Policies and Procedures." (Issued on January 20, 1998. Withdrawn September 25, 2018.)
As seen above and as presented in Attachment 1, new § 812.35(a) requires prior FDA approval for changes to the investigational device, including manufacturing changes, that constitute a significant change in design or basic principles of operation, or are not made in response to information gathered during the course of an investigation. Additionally, changes to the protocol that affect the validity of the results from the study, the risk to benefit relationship, the scientific soundness of the investigational plan, or the rights, safety or welfare of the subjects also require prior approval. On the other hand, changes to the investigational device (including manufacturing changes) and changes to the clinical protocol can be made without prior approval if the changes do not fall into any of the above categories and the IDE sponsor reports the modifications to the agency within 5 days of implementation. (Hereafter, this type of submission will be referred to as a "5-day notice.") Finally, under the new regulation, the agency codified its current policy of allowing minor changes to the investigational plan to be made via notification in the sponsor’s annual report to the IDE. FDA is providing this guidance to restate the information contained in the preambles of the proposed and final rules to help clarify those types of changes that require prior approval versus those that can be reported in a 5-day notice or in the annual progress report. In addition, guidance is provided on the preparation and review procedures for industry and FDA staff, respectively, for each of these types of submissions.
III. CHANGES REQUIRING PRIOR APPROVAL VS. CHANGES SUBJECT TO 5-DAY NOTICE
Although the new statute and its implementing regulation identify certain criteria that must be satisfied in order for a change to be effected without prior agency approval, these criteria are fairly general. That is, device/manufacturing and protocol changes that could meet the criteria range from the very minor to the more complex. For example, device modifications could range from a packaging change to a change in material. Similarly, protocol changes may involve changes to the follow-up schedule for subjects or may involve the use of new primary/secondary endpoints. § 812.35(a)(3)(iii) of the regulation describes the types of credible information that sponsors should use to determine which changes could be implemented without prior approval from those that require prior approval. To assist sponsors in deciding if a change may be considered under a 5-day notice, potential changes to the device/manufacturing process and the protocol, as well as the supporting credible information for these changes, are discussed below.
A. CHANGES TO THE DEVICE OR THE MANUFACTURING PROCESS
Under the new law, certain developmental changes to the investigational device, including manufacturing changes, are eligible for implementation during the course of the clinical investigation without prior FDA approval. Modifications that constitute a significant change in design or basic principles of operation, or that were not made in response to information gathered during the course of an investigation, however, may not be made without prior approval of an IDE supplement. In the guidance document entitled, "Deciding When to Submit a 510(k) for a Change to an Existing Device,"2 the agency identified generic types of device and manufacturing modifications. Although this guidance applies to modifications of marketed devices, the types of changes identified are also applicable to investigational devices. These include changes to the control mechanism, principle of operation, energy type, environmental specifications, performance specifications, ergonomics of patient-user interface, dimensional specifications, software or firmware, packaging or expiration dating, sterilization, and the manufacturing process (including the manufacturing site).
In the preamble of proposed IDE Modification regulation, FDA stated that all changes to the basic principles of operation of a device would be considered significant changes and solicited comments on this premise. No comments were received. Therefore, in the final rule, FDA stated that all changes to the basic principles of operation of a device should be submitted in an IDE supplement for prior approval.
For the remaining types of device and manufacturing changes listed above, the changes can range from minor to significant, depending upon the particular device, the type of modification, and the extent of the modification. According to the statute, it is the sponsor’s responsibility to determine if a change made to the device or the manufacturing process would be considered a significant change requiring prior agency approval. To help sponsors determine if a change represents a significant change, the decision tree in Attachment 2 may be used in the decision-making process. According to the decision tree, the sponsor should first conduct a risk analysis to help identify the potential risks that the change to the device and/or manufacturing process may present. If the risk analysis identifies a new type of risk, then prior approval would be needed. If, however, no new type of risk is identified, the change may be eligible for implementation under a 5-day notice, but the sponsor should confirm this through the use of credible information.
Following the flowchart, FDA recommends that the sponsor use the data generated by design control procedures or other credible information to help determine whether the change has a significant affect on the device design. Credible information that may be used to support developmental changes in the device (including manufacturing changes) is defined in the regulation under § 812.35(a)(3)(iii)(A). Credible information may include data generated under the design control procedures of § 820.30, pre-clinical/animal testing, peer reviewed published literature, or other reliable information gathered during a trial or marketing. Any of these types of information may be used to support a design and/or manufacturing change, meeting the criteria discussed above, in a 5-day notice.
To help illustrate this decision-making process, consider a change in material from polyvinylchloride (PVC) to silicone in a central venous catheter. In accordance with the decision tree, the sponsor would conduct the risk analysis. The risk analysis would assess the impact of this change on the safety and effectiveness of the device. The analysis may determine that this change may impact the device’s strength, flexibility and biocompatibility compared to the unmodified device, but not lead to the identification of any new risks. The sponsor would then proceed to evaluate the above risks using credible information. This may include conducting appropriate bench and animal testing to determine that the change does not significantly alter the device’s strength, flexibility or biocompatibility. As a part of these activities, the sponsor should also conduct any other testing that may have been identified to the sponsor in an FDA-issued guidance document, disapproval letter, or conditional approval letter for this device and that is relevant to the change. If the results of the testing demonstrate that all of the risks (those identified in the risk analysis and those identified by the agency in its previous correspondence to the firm) have been adequately addressed, then the change could be implemented without prior FDA approval.
Using the same device in a second example, consider a change in the diameter of the lumen of the catheter. If no new types of risks are identified in the risk analysis, the manufacturer could proceed to conduct the verification and validation testing, as appropriate. If the testing demonstrates that the design input requirements are met, the change could be implemented without prior FDA approval. If, however, during the testing, it is determined that the intended flow rate was compromised by the change in diameter, then the manufacturer would have two options. The manufacturer could adjust the modification so that the original intended flow rate is still achieved or the manufacturer could submit an IDE supplement, including a justification for the change, and pursue FDA approval of the modified device.
By using the data generated by design control procedures or other credible information, the manufacturer should be able to identify significant changes to the investigational device or manufacturing process. It should be noted that in the preamble to the final rule, the agency stated that changes intended to enhance significantly the safety or effectiveness of the device may be implemented without prior approval, if the developmental changes do not constitute a significant change in device design. Thus, although significant changes in design are not permitted under this new provision, changes that could have a significant effect on safety or effectiveness are permitted as long as they do not represent significant design changes. Below are some examples of device design/manufacturing changes that have been implemented through the 5-day notice provision:
A modification to the delivery system for a stent to reduce the shaft outer diameter so it could be used in a smaller sized catheter sheath introducer and therefore permit a smaller vascular access site.
A design modification to the tip of the catheter used during stent placement to reduce the risk of the tip being snagged on the stent strut when it is being withdrawn.
A change in an adhesive was implemented for an invasive device.
For a pacemaker, a change was made to the programmed mode that the device was shipped in to the mode that is most commonly used by the clinical investigators.
A marker band was added to an investigational device to enhance visualization during fluoroscopy.
The shelf life of a device was extended from 6 months to two years.
2 This guidance may be found at "Deciding When to Submit a 510(k) for a Change to an Existing Device".
B. CHANGES TO THE CLINICAL PROTOCOL
Changes or modifications to the clinical protocol may be reported in a 5-day notice if the changes do not affect: a) the validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; b) the scientific soundness of the investigational plan; or c) the rights, safety, or welfare of the human subjects involved in the investigation. The sponsor is responsible for initially determining if the change meets the statutory criteria. This determination should be made by the person in the company responsible for such decisions, and should be based on the agency’s definition of credible information. Under § 812.35(a)(3)(iii)(B), credible information is defined as the sponsor’s documentation supporting the above conclusions and may include information such as peer reviewed published literature, the recommendation of the clinical investigator(s), and the data gathered during the clinical trial or marketing. In addition, as stated in the preamble to the final rule, credible information may include IRB approval and/or concurrence of the data and safety monitoring board (DSMB). In general, protocol modifications that serve to increase patient safety would meet these statutory criteria. Thus, the following types of changes should be appropriate for implementation without prior FDA approval:
- Increasing the frequency at which data or information is gathered or lengthening the subject follow-up period. For example, modifying the follow-up schedule such that subjects return every month for evaluation rather than every 2 months or extending the follow-up period from 6 months to one year.
- Modifying the protocol to include additional patient observations or measurements. For example, adding a quality of life assessment or performing additional tests on previously collected subject specimens.
- Modifying the inclusion/exclusion criteria to better define the target patient population. For example, modifying the criteria to exclude subjects who had been exposed to another investigational device within a certain time period prior to participation in the current study.
- Modifying the secondary endpoint(s). For example, eliminating the assessment of post-void residuals in a benign prostatic hyperplasia (BPH) study if this secondary endpoint does not represent a clinically significant outcome measure.
Alternatively, the following types of protocol modifications can have a significant effect on the validity of the data resulting from the trial and/or on the scientific soundness of the trial design and thus would not generally be eligible for implementation without prior approval.
- Change in indication. For example, narrowing the indication to a subgroup upon which the device appears to be working better than in the overall population. Such a change could lead to a Type I error in the subgroup and a lack of power to evaluate the subgroups in general.
- Change in type of study control. For example, a change from an active control (legally marketed device) to the use of historical (literature) control.
- Change in the primary endpoint variable. For example, deciding mid-study to identify a new primary endpoint when the endpoint had not been included in the original protocol and thus data was not being collected to evaluate it. As a second example, exchanging the primary and secondary endpoints because it does not appear that the primary endpoint will meet the success criteria. Such changes would most likely require statistical adjustments, e.g., the original sample size estimate may no longer be valid. Alternatively, adding a new endpoint to the original study endpoints may not require prior approval if no new risks to the patient are introduced when collecting the data to evaluate the endpoint.
- Reduction in sample size. Such a change would normally lead to a loss in statistical power.
- Change in the method of estimation. For example, changing from an exact method (e.g., hypergeometric model) to an approximate method (e.g., Chi Square).
- Early termination of the study (except for reasons related to patient safety). Early termination may invalidate the data as early results may not be typical. For example, deciding that a 6-month rate of ventricular tachycardia recurrence rather than the planned 1-year rate will suffice because few recurrences have occurred at 3 months may later turn out to be an invalid assumption.
A change to the protocol to increase the sample size or expand the number of investigational sites continues to require submission and approval of an IDE supplement. FDA believes that expanding the study to increase either the number of subjects exposed to an investigational device or to increase the number of institutional sites participating in the trial affects the rights, safety, or welfare of the subjects and thus may not be implemented under the 5-day notice provision.
Below are some examples of protocol changes that have been implemented through the 5-day notice provision:
A modification to the inclusion/exclusion criteria to make the study population consistent with the intended target patient population once the device is approved and to more closely match that being studied in the European clinical trial.
A change to the protocol to allow the use of a 6 French or greater guide catheter rather than the 7 French or greater that was identified in the original protocol.
A modification to include the use of a commercially available device to insure that pacing therapy would be available if the patient connection cable failed or there was a poor connection. That is, the sponsor added a backup safety feature to the study design.
The above discussion of the types of device/manufacturing and protocol changes that may be implemented as a 5-day notice could not be exhaustive due to the range of investigational devices and modifications that could potentially be made to the investigational plan. FDA recommends that if an IDE sponsor is uncertain whether a proposed change meets the statutory criteria, the sponsor contact the reviewing division.
In addition to the considerations identified above, sponsors who have entered into an agreement and/or a determination with the agency under sections 520(g)(7)(A) or 513(a)(3)(D)(i) of the act with regard to the investigational plan or the data needed to demonstrate effectiveness of the investigational device should consider whether the proposed protocol or device change may invalidate the agreement or determination. If an agreement or determination is in effect, FDA recommends that the IDE sponsor contact the reviewing division to discuss the proposed change and any impact it may have on the agreement or determination before the change is implemented.
Finally, when considering the effect that a change to the device, the manufacturing process, or the clinical protocol may have, the sponsor should consider if the poolability of the resulting clinical data would be affected. Sponsors should be prepared to justify why such changes did not affect the validity of the resulting data at the time of the submission of the marketing application.
IV. OTHER CHANGES TO THE INVESTIGATIONAL PLAN
Under § 812.35(a)(4), minor changes to the purpose of the study, the risk analysis, monitoring procedures, labeling for the investigational device, informed consent materials, and IRB information may continue to be submitted in an IDE annual report if the changes do not affect: (i) the validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; (ii) the scientific soundness of the investigational plan; or (iii) the rights, safety, or welfare of the human subjects involved in the investigation. As in the case of protocol modifications, the types of changes that would normally satisfy these criteria would be those that would serve to increase patient safety, e.g., clarifying the instructions for use, providing additional information in the informed consent document, or enhancing the monitoring procedures. Below, each of these parts of the investigational plan is discussed and specific examples are provided to illustrate the types of changes that would usually be considered appropriate for submission in an annual report.
A. Purpose.
According to § 812.25(a), the purpose of the study includes the name and intended use of the device as well as the objectives and duration of the investigation. Examples of changes that may be made to this section of the investigational plan and reported in the annual report include:
- Changes to the name of the device. This type of change may be made provided that the new name does not imply a new intended use. Name changes that are made in conjunction with a modification to the device, however, should be submitted with the device modification.
- Clarifications to the intended use of the device. Such changes may be made if the modifications do not implicitly or explicitly affect the intended use.
- Minor modifications to the study objectives. These may be of several types:
- The study objectives may be revised to provide clarification as long as the intent of the objectives and the study endpoints are not changed.
- Study objectives related to future labeling claims for the device may be added if such changes meet the statutory criteria. If, however, the change in the objectives requires a protocol modification, the change should be submitted as an IDE supplement or within 5 days of implementation, as appropriate for the protocol modification.
- Changes in the duration of the investigation. If the investigation will take less time or more time to complete than was anticipated at the time the application was submitted, this information should be submitted in the annual report.
B. Risk Analysis.
If information to be added to the risk analysis does not affect the risk to benefit relationship, it may be reported in the annual report. During the course of the investigation, if the sponsor becomes aware of information that may adversely affect the risk analysis, however, this information should be submitted in a supplement indicating that the risk to benefit relationship has changed.
C. Monitoring Procedures.
A change in the name and/or address of the monitor may be made without prior approval and submitted in the annual report. In addition, changes in the monitoring procedures that are consistent with the "Guideline for the Monitoring of Clinical Investigations" are eligible for this type of reporting mechanism.
D. Labeling.
Labeling changes that clarify the instructions for use or serve to increase subject safety may be submitted in the annual report. Adding contraindications, hazards, adverse effects, interfering substances/devices, warnings, or precautions to the labeling, however, may require concomitant changes to the protocol (e.g., modifications to the exclusion criteria) and thus should be submitted as an IDE supplement or within 5 days of implementation, as appropriate for the protocol modification.
E. Informed Consent.
Revisions to the informed consent materials may be made without prior approval and submitted in the annual report if the changes are, for example, to include preliminary results from the trial (if in agreement with expected outcome(s)), clarify the risks and/or potential benefits of the investigational device, clarify the procedures/tests to which the subjects may be subjected, etc.
F. IRB information.
A change in the IRB chairperson or address should be reported in the annual report. Changes in approval status of the study, however, must be reported in accordance with § 812.150(b)(2).
V. PROCEDURES
As discussed above, new § 812.35(a) provides for three approval/notification mechanisms for changes or modifications that may occur during the course of a clinical investigation. Below, the sponsor’s responsibilities in the various types of submissions and FDA’s actions on them are discussed.
A. CHANGES REQUIRING PRIOR APPROVAL
Certain changes or modifications to a clinical investigation require submission of an IDE supplement and approval by FDA before implementation. Prior approval is required for changes to the device (including manufacturing changes) that constitute a change in the basic principles of operation or a significant change in design or changes that were not made in response to information gathered during the course of the investigation. Additionally, prior approval is required for changes to the investigational plan that affect the validity of the data resulting from the study, the risk to benefit relationship for subjects enrolled in the study, the scientific soundness of the investigation, or the rights, safety or welfare of subjects.
IDE supplements requesting these changes should include a detailed description of the change (cross-referenced to the appropriate sections of the original submission), an explanation of why the change is being requested, an assessment of the impact of the change on the study and documentation supporting the change. The supporting documentation needed depends on the change being requested. It may include preclinical bench/animal testing, peer reviewed published literature, risk analysis of the change, statistical analysis of the impact on the study, etc.
As with all IDE supplements, the submission should reference the IDE number and be submitted in triplicate to:
U.S. Food and Drug Administration
Center for Devices and Radiological Health
Document Mail Center – WO66-G609
10903 New Hampshire Avenue
Silver Spring, MD 20993-0002
FDA will review these submissions within 30 days and issue an approval, conditional approval, or disapproval letter.
As appropriate, and in accordance with §§ 56.110 and 56.111, changes submitted to FDA for prior approval may also require approval from the participating IRBs prior to implementation. At a minimum, the IRBs should be notified of such changes in order to be kept fully informed.
B. CHANGES EFFECTED WITH NOTICE TO FDA
Device/manufacturing and protocol changes under § 812.35(a)(3) may be implemented without prior FDA approval if a notice of the change is submitted to the IDE not later than 5 working days after making the change. Implementation of changes to a device are considered to occur on the date the device, manufactured incorporating the design or manufacturing change, is distributed to investigator(s). Changes to the protocol are considered to occur when the sponsor notifies a clinical investigator that the change should be implemented in the protocol. For a sponsor-investigator study, the change to the protocol is considered to occur when the sponsor-investigator incorporates the change in the protocol.
Notices should be clearly identified as a "Notice of IDE Change" and be submitted in triplicate, referencing the IDE number, to the above address. FDA will review the Notice of IDE Change within 30 days and, normally, there will be no response from FDA to the sponsor. If clarification or additional information is needed, the agency will generally request this information by telephone.
While the statute and the regulation clearly state that it is the sponsor’s responsibility to determine if a device, manufacturing, or protocol change meet the statutory criteria for implementation without prior agency approval, under § 812.35(a)(3)(v), FDA reserved the right to question the sponsor’s determination. If the agency has reason to believe, based on the information submitted in the Notice of IDE Change or on other available information, such as reports of adverse events, that the modification did not meet the statutory criteria, FDA will notify the sponsor that the change should have been reviewed and approved before being implemented. FDA recognizes the potential impact that this action could have on the IDE sponsor and the clinical trial and, therefore, intends to take such action only if the agency determines that the modification could jeopardize patient safety, the scientific soundness of the investigation, or the validity of the data resulting from the trial. Such determinations will be made by the individuals authorized to approve IDEs.
Changes submitted to FDA in a Notice of IDE Change should be reported to the participating IRBs in order to keep them fully informed. In general, IRB approval should not be required, as the changes presumably do not affect the rights, safety, or welfare of the subjects. There may be cases, however, in which, depending upon the type of protocol change being implemented, IRB approval may be required under § 56.110.
The information required in a Notice of IDE Change varies depending upon whether the change is a developmental change in the device or manufacturing process or a change to the clinical protocol. The requirements for these types of submissions are discussed below. The FDA reviewer checklist for a Notice of IDE Change summarizes this information and is provided in Attachment 3.
1. Developmental Changes
A Notice of IDE Change may be submitted for developmental changes in the device (including manufacturing changes) that do not constitute a significant change in design or basic principles of operation and that are made in response to information gathered during the course of the investigation. The notice should include: a) a summary of the relevant information gathered during the course of the investigation upon which the change was based; b) a description of the change to the device or manufacturing process (cross-referenced to the appropriate sections of the original device description or manufacturing process), and c) documentation of the credible information to support the change.
According to the IDE Modification regulation (§ 812.35(a)(3)(iii)(A)), credible information for developmental changes includes data generated under the design control procedures of § 820.30, preclinical/animal testing, peer reviewed published literature, or other reliable information such as clinical information gathered during a trial or marketing. If design controls are used to assess the change, the documentation submitted in the notice should include a statement that no new risks were identified by an appropriate risk analysis and that the verification and validation testing, as appropriate, demonstrate that the design outputs met the design input requirements. If preclinical/animal testing is used to assess the change, documentation should include information to indicate that the appropriate testing was conducted to address safety and performance concerns (for example, to meet a standard that is identified as a device input requirement). If peer reviewed published literature is used to assess the change, copies of the published literature should be provided.
It is important to note that the device/manufacturing change should not be implemented before the credible information has been generated to assess the proposed change. Similarly, the evaluation and/or testing performed to assess the change must be completed prior to submission of the 5-day notice to the agency. One of the most common problems with the implementation of this new provision has been that IDE sponsors have submitted their notices before the testing was conducted and instead submitted their proposed testing or a promissory note to conduct the testing. The agency is taking this opportunity to remind sponsors that the assessment of the change and any supporting testing must be completed before the change is implemented, and the Notice of IDE Change must be submitted to the agency within 5 days of implementation.
2. Changes to the Clinical Protocol
A Notice of IDE Change may be submitted for changes to the clinical protocol that do not affect: a) the validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; b) the scientific soundness of the investigational plan; or c) the rights, safety, or welfare of the human subjects involved in the investigation. A notice for a protocol change should include: 1) a description of the change (cross-referenced to the appropriate sections of the original protocol); 2) an assessment supporting the conclusion that the change does not have a significant impact on the study design or planned statistical analysis; and 3) a summary of the information that served as the credible information supporting the sponsor’s determination that the change does not affect the rights, safety or welfare of the subjects.
According to the IDE Modification regulation (§ 812.35(a)(3)(iii)(B)), credible information to support changes to the clinical protocol is defined as the sponsor’s documentation supporting the conclusion that a change does not have a significant impact on the study design or planned statistical analysis, and that the change does not affect the rights, safety, or welfare of the subjects. Documentation may include information such as peer reviewed published literature, the recommendations of the clinical investigator(s), and/or the data generated during the clinical trial or marketing. As previously stated, FDA would also consider IRB approval or concurrence of the DSMB to serve as credible information to support the protocol change.
C. CHANGES SUBMITTED IN AN ANNUAL REPORT
Certain other changes to the investigational plan may be reported to FDA in an IDE annual report. Minor changes eligible for reporting in an annual report are those that would not affect: a) the validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; b) the scientific soundness of the investigational plan; or c) the rights, safety or welfare of the subjects involved in the investigation. As discussed above, these could include minor changes to the purpose of the study, risk analysis, monitoring procedures, labeling, informed consent materials and institutional review board information.
Sponsors should follow "The Suggested Format for IDE Progress Report" (see Attachment 4) in preparing this submission. The report should describe the changes that have been made and the reason for the changes. The submission should be identified as an IDE Annual Report and be submitted in triplicate, referencing the IDE number, to the above address.
Normally there will be no response from FDA to the sponsor for these types of changes. If clarification or additional information is needed, FDA may request this information by telephone or letter.
Changes submitted to FDA in an annual report should also be reported to the participating IRBs. IRB approval should not be required as the changes presumably do not affect the rights, safety, or welfare of the subjects, however, notification of such changes is required to keep the IRBs fully informed.
VI. CONCLUSION
Under § 812.35(a) of the IDE regulation, it is the sponsor's responsibility to consider the effect that any change made to the investigational plan may have on the clinical investigation and the resulting data. Any change to the basic principles of operation of a device is considered to be a significant change and, thus, requires prior FDA approval. In assessing the effect of a device design and/or manufacturing change, a risk analysis and supporting credible information should help to identify those changes that represent a significant change. For a protocol change, the sponsor should assess, using credible information, whether the change affects: a) the validity of the data or information resulting from the completion of the approved protocol, or the relationship of likely patient risk to benefit relied upon to approve the protocol; b) the scientific soundness of the investigational plan; or c) the rights, safety or welfare of the human subjects involved in the investigation.
This guidance document incorporates the discussion from the preambles of the proposed and final rules amending the IDE regulation to implement new section 520(g)(6) of the act. Because this section of the act permits sponsors to implement certain changes to the device/manufacturing process and the clinical protocol without prior approval, the agency considers this provision to be part of Congress’ intent to reduce regulatory burden during device development. Implementation of this provision is, therefore, an important part of agency’s least burdensome approach to device regulation, and FDA encourages industry to take advantage of this new mechanism.
Attachment 1
|
Type of Change |
Type of Submission |
||
|---|---|---|---|
| Supplement | 5-Day Notice | Progress Report | |
| I. Device/Manufacturing Changes | |||
| Not made in response to information from the investigation | |||
| Made in response to information from the investigation and is | |||
| A significant change in design or any change to the basic principles of operation | |||
| Not a significant change in design/manufacturing | |||
| II. Protocol Changes | |||
| Affects validity of data/information; patient risk to benefit relationship; scientific soundness of plan; or rights, safety or welfare of subjects | |||
| Does not affect validity of data/information; patient risk to benefit relationship; scientific soundness of plan; or rights, safety or welfare of subjects | |||
| III. Minor Investigational Plan Changes | |||
| Does not affect validity of data/information; patient risk to benefit relationship; scientific soundness of plan; or rights, safety or welfare of subjects | |||
| Purpose of study | |||
| Risk analysis | |||
| Monitoring procedures | |||
| Labeling | |||
| Informed consent materials | |||
| IRB information | |||
| Does affect validity of data/information; patient risk to benefit relationship; scientific soundness of plan; or rights, safety or welfare of subjects | |||
| Purpose of study | |||
| Risk analysis | |||
| Monitoring procedures | |||
| Labeling | |||
| Informed consent materials | |||
| IRB information | |||
Attachment 2
Proposed Changes to the Device or the Manufacturing Process
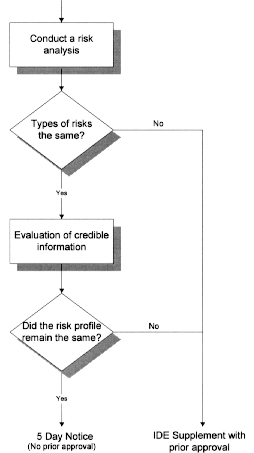
* The credible information should include all tests required by FDA during the approval process for the original IDE, if relevant to the change.
Attachment 3
 |
DEPARTMENT OF HEALTH & HUMAN SERVICES |
Public Health Service Food and Drug Administration |
| Memorandum |
NOTICE OF IDE CHANGE
Date: ______________________________
File: ______________________________
Reviewer: ______________________________
This IDE supplement submission contains modifications to the device design, manufacturing process, and/or protocol allowable under 21CFR812.35 and the sponsor is providing notice of these changes within five working days of implementation.
Description of Modification(s):
_____________________________________________________________
_____________________________________________________________
_____________________________________________________________
_____________________________________________________________
_____________________________________________________________
|
Design |
Manuf. Change |
|||||
|---|---|---|---|---|---|---|
|
Yes |
No |
Yes |
No |
|||
| 1. | Is there a change to the device design or manufacturing process? If no, go to number 3. |
 |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
||
|
|
|
|
|
||
| 2. |
Is a summary provided of the credible information that served to support the change? (Credible information may include a summary of the information generated under the design control procedures of Sec. 820.30, preclinical/animal testing, peer reviewed published literature, or other reliable information such as clinical information gathered during a trial or marketing (outside the U.S.)) |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
||
|
|
|
||||
|
Yes |
No |
|||||
| 3. | Is there a change to the clinical protocol? | |
|
|||
|
|
|
||||
|
|
|
||||
|
|
|
||||
|
|
|
||||
| 4. |
Is a summary provided of the credible information supporting the change? (Documentation includes information such as peer reviewed published literature, the recommendation of the clinical investigator(s), the data gathered during the clinical trial or marketing, IRB approval, and/or DSMB concurrence.) |
|
|
|||
|
|
|
||||
|
|
|
||||
Attachment 4
Suggested Format for IDE Progress Report
I. The Basics
- IDE Number
- Device name and indication for use
- Sponsor's name, address and phone number
- Contact person
II. Study Progress
(Data from beginning of the study should be reported, unless otherwise indicated.)
- Brief summary of study progress in relation to investigational plan
- Number of investigators/investigational sites (attach list of investigators)
- Number of subjects enrolled (by indication or model)
- Number of devices shipped
- Brief summary of results
- Summary of anticipated and unanticipated adverse effects
- Description of any deviations from the investigational plan by investigators (since last progress report)
III. Risk Analysis
- Summary of any new adverse information (since last progress report) that may affect the risk analysis; this includes preclinical data, animal studies, foreign data, clinical studies, etc.
- Reprints of any articles published from data collected from this study
- New risk analysis, if necessary, based on new information and on study progress
IV. Other Changes
- Summary of any changes in manufacturing practices and quality control (including changes not reported in a supplemental application)
- Summary of all changes in investigational plan not required to be submitted in a supplemental application
V. Future Plans
- Progress toward product approval, with projected date of PMA or 510(k) submission
- Any plans to change investigation, e.g., to expand study size or indications, to discontinue portions of the investigation or to change manufacturing practices (NOTE: Actual proposals for change should be made in a separate supplemental application)
Submit Comments
You can submit online or written comments on any guidance at any time (see 21 CFR 10.115(g)(5))
If unable to submit comments online, please mail written comments to:
Dockets Management
Food and Drug Administration
5630 Fishers Lane, Rm 1061
Rockville, MD 20852
All written comments should be identified with this document's docket number: FDA-2020-D-0957.