Preparation and LC/MS/MS Analysis of Honey for Fluoroquinolone Residues
September 29, 2006
Method Developed by
Florida Department of Agriculture and Consumer Services
-
Title
Preparation and LC/MS/MS Analysis of Honey for Fluoroquinolone Residues
-
Principle
Honey is dissolved in water, and then any fluoroquinolone residues are extracted into acetonitrile after acidification. Water is removed from the acetonitrile by passing the extract through a sodium sulfate column. The extract is concentrated, then assayed using LC-MS/MS.
-
Scope
This method will allow the determination and confirmation of enrofloxacin and ciprofloxacin in honey at concentrations above 2.5 ppb.
- Materials and Apparatus, Media and Reagents, Associated Documents
- 4.1 Materials and Apparatus
- Vortex: Thermolyne model 65800 or equivalent
- Evaporator: Zymark TurboVap LV or equivalent
- Centrifuge: Jouan model GR 20 22, IEC model C 6000, ThermoElectron Corp model 3L GP 2500 or equivalent
- Pipettes: Rainin electronic pipettes, fisherbrand disposable transfer pipettes catalog #137117 or equivalent
- Solid phase extraction cartridge: sodium sulfate with polypropylene Membrane, Whatman, cat # 6805-8020
- Syringes: 20 mL slip tip, latex free
- 50 mL centrifuge tube: polypropylene, Fisher, cat. # 05-539-9
- Autosampler vials, amber: National Scientific Company, Target I-D vials, cat. # C4011-2W
- Volumetric glassware: 5 mL
- 4.2 Reagents
- Acetonitrile, Fisher Optima grade
- Water, deionized - Milli-Q 16 megaohm or better
- Glacial acetic acid, HPLC grade
- Formic acid, 88% or better
- NaCl, certified ACS
- 4.1 Materials and Apparatus
- Specific Procedures
-
5.1 Safety Precautions
All work with organic solvents must be done in a chemical fume hood. Personal protective gear is required, including safety glasses, solvent resistant gloves and laboratory coat.
-
5.2 Cross Contamination
Disposal glassware is used wherever possible to eliminate the possibility of cross contamination. Great care must be taken to clean non disposable glassware in the procedure.
5.3 Reagent Preparation
- 2% acetic acid
- Place 20 mL of acetic acid in a 1000 mL graduated cylinder.
- Add 980 mL of deionized water.
- Mix by inversion.
- 16/84 acetonitrile/2% acetic acid
- Place 160 mL of acetonitrile in a 1000 mL graduated cylinder.
- Add 840 mL of 2% acetic acid solution.
- Mix by inversion.
- HPLC Mobile Phase:
- Formic acid in water (0.1%)
- Add 1 mL of 88 - 100 % formic acid to a 1000 mL graduated cylinder using a volumetric pipette or suitable electronic pipette.
- Dilute to a final volume of 1000 mL with deionized water and mix well by inversion.
- Formic acid in acetonitrile (0.1%)
- Add 1 mL of 88 - 100 % formic acid to a 1000 mL graduated cylinder using a volumetric pipette or suitable electronic pipette.
- Dilute to a final volume of 1000 mL with acetonitrile and mix well by inversion.
Note: Other quantities of these reagents may be prepared by scaling the proportions appropriately.
- Formic acid in water (0.1%)
- 2% acetic acid
-
5.4 Sample Extraction
- Weigh 2.5 grams (± 0.03 grams) of honey into a 50 mL disposable centrifuge tube (tube A). Record this weight.
- Add 5 mL of deionized water to the honey sample in tube A.
- Prepare a reagent blank sample by measuring 7.5 mL of deionized water into a 50 mL disposable centrifuge tube (tube A).
- Vortex mix and/or hand shake mix until all of the honey dissolves into the water.
- Add 10 mL of acetonitrile to the sample in tube A.
- Add 200 µL acetic acid.
- Vortex mix for approximately 30 seconds.
- Add 2 grams (± 0.2grams) of NaCl.
- Vortex for approximately 15 seconds.
- Centrifuge at 2400 RCF for 5 minutes.
- Attach a Sodium Sulfate cartridge to a 20 mL disposable syringe and place inside a clean 50 mL disposable centrifuge tube (tube B).
- Using disposable pipettes, transfer the acetonitrile layer (top layer) from tube A to the 20 mL syringe attached to the sodium sulfate cartridge. Sample may begin to gravity feed through the cartridge at this point and collect inside tube B. It is important that the syringe/drying cartridge assemble remain inside tube B so that no sample is lost.
- Add 10 mL of acetonitrile to the 50 mL disposable centrifuge tube (tube A).
- Vortex mix for approximately 30 seconds.
- Centrifuge at 2400 RCF for 5 minutes.
- Repeat step 12.
- Apply pressure using the syringe plunger and pass the acetonitrile extract through the sodium sulfate cartridge into the clean 50 mL disposable centrifuge tube (tube B).
- Concentrate the extract in tube B to less that 1 mL in the turbovap at 55 degrees centigrade with nitrogen.
- Quantitatively transfer the extract in tube B to a 5 mL volumetric flask and dilute to volume with 16/84 acetonitrile/2% acetic acid in water.
- Mix sample in flask by inversion.
- Remove a portion of the sample from the volumetric flask and place in an amber flat bottom autosampler vial.
- Sample is now ready for analysis by LC-MS/MS.
-
5.5 LC/MS/MS Instrumental Analysis
- Instrument: Thermo Finnigan Linear Ion Trap LC/MS/MS with Agilent 1100 HPLC. See attachment A for the instrument method.
- Mobile Phase: solvent A = H2O w/0.1% formic acid, solvent B = acetonitrile with 0.1% formic acid.
- Gradient Conditions:
Time 0 min: 95% A, 5% B flow = 200 µL/min Time 15 min: 5% A, 95% B flow = 200 µL/min Time 25 min: 5% A, 95% B flow = 200 µL/min Time 25.1 min: 95% A, 5% B flow = 500 µL/min Time 30.0 min: 95% A, 5% B flow = 500 µL/min Time 30.1 min: 95% A, 5% B flow = 200 µL/min Time 32.1 min: 95% A, 5% B flow = 200 µL/min - Injection Volume 10 uL. Autosampler at ambient temperature.
- Column: Waters/2.0x150, XTerra phenyl / 5 micron. Column heater 40°C isothermal.
- LTQ Method: Run time = 32.1 minutes. Divert valve: 0 min to waste, 5.05 min to source, 15.05 min to waste.
- Selected ions and retention times: Positive Mode Electrospray LC/MS/MS
Ciprofloxacin:Enrofloxacin:
332>288 transition in MS/MS (used for quantification) 332>288>268 transition in MS^3 (major qualitative ion transition) 332>288>245 transition in MS^3 (major qualitative ion transition) Retention time (approximate): 10.5 minutes
360>316 transition in MS/MS (used for quantification) 360>316>245 transition in MS^3 (major qualitative ion transition) 360>316>288 transition in MS^3 (minor qualitative ion transition) Retention time (approximate): 10.9 minutes
Note: MS/MS on the ion trap yields one primary fragment ion and some smaller less abundant peaks for both ciprofloxacin and enrofloxacin. An additional stage of MS analysis (MS^3) is performed on the primary fragment ion from the MS/MS experiment and yields sufficient points for confirmation.
- Tune File: See example in Attachment B (STATESCREEN_122804)
-
Processing Files: Three to five level linear curves are used for quantification. See Attachment C for example of an extracted ion chromatogram. (FLUROQUINOLONES_CURVE1, FLUROQUINOLONES_CURVE2, FLUROQUINOLONES_CURVE3, FLUROQUINOLONES_CURVE4)
Note: Other processing files may be needed based on the type of curve, number of points, or type of calibration performed.
-
5.6 Quantification
- The MS/MS transition (332>288 for ciprofloxacin and 360>316 for enrofloxacin) are used for quantification.
- Reference standards run before and after (bracket) the samples in a given sequence shall have detector response variations no greater than 20% relative percent difference. See Attachment D for an example of a run sequence. Standards are injected before and after a group of 5 - 10 samples, typically at the beginning, middle and end of a run. If the detector response of standards injected at the beginning and end of an analytical run vary by more that 20%, samples may be quantitated using the standards in the middle of the run. Samples must be reinjected if detector response drifts greater than 20% are encountered.
-
Calibration curves with standard responses that bracket that of the sample response are routinely used. Standards are prepared at five concentrations which vary depending on the concentration of analyte in the samples but usually corresponding to 2.5, 5.0, 10, 25 and 50 ppb. At least four standards must be used to define a curve. A dilution factor of 2 may be used to convert the analyte concentration in the extract to the concentration of the analyte in the sample by weight.
Alternately, standard concentrations that represent the equivalent sample ng/g level of the target compound may be utilized in the data software with a dilution factor of 1.0. The curve is a linear fit, not forced through zero. If needed, a 1/x weighting may be used to more accurately quantitate low concentrations. Acceptable curves have correlation coefficients of 0.99 or greater. All quantitative sample responses must fall within the calibration curve.
- Samples exceeding the highest reference standard shall be diluted within the standard curve using a blank matrix.
- Attachment E provides information on the use of standard additions to calculate the concentration if matrix effect of the honey are causing poor quantitation. Please note that quantitation using standard additions has not been fully validated by the method developer.
-
5.7 Standards
Individual standards of enrofloxacin and ciprofloxacin are prepared in a solution of 16/84% acetonitrile/acetic acid (2%) by the standards section. These standards are each prepared at levels of 0.05, 0.1, 0.2, 0.5, and 1.0 ng/uL. These standards are stored in amber disposable vials in the freezer at -10°C. Standards should be prepared fresh for each batch of analyses.
Preparation of the standards for analysis of honey samples is as follows:
- Remove a single vial of the stock solution for each compound, at each level, from the freezer and allow standards to come to room temperature.
-
Place 125 µL of the 0.05 ng/ µL ciprofloxacin and 125 µL of the 0.05 enrofloxacin standard in a 5 mL volumetric flask. Dilute to volume with honey extract*. This is the working mixed standard at 1.25 ng/mL that corresponds to a target level for enrofloxacin and ciprofloxacin in the sample of 2.5 ng/g for each compound. Discard the remaining standards in the original stock standard vials removed from the refrigerator. See note - Section 5.7.7.
* Note - honey extract is a final extract (i.e. ready for instrumental analysis). It is generated by taking negative control honey and extracting it per this SOP. The reference matrix utilized for the method validation was tupelo honey.
- Repeat step 2 for the remaining standard concentrations of 0.1, 0.2, 0.5, and 1.0 ng/µL standards.
-
The above steps provide the following working mixed standards for analysis and calibration of ciprofloxacin and enrofloxacin.
1.25 ng/mL mixed working standard-in-matrix (2.5 ng/g sample equivalent)
2.5 ng/mL mixed working standard-in-matrix (5.0 ng/g sample equivalent)
5.0 ng/mL mixed working standard-in-matrix (10.0 ng/g sample equivalent)
12.5 ng/mL mixed working standard-in-matrix (25.0 ng/g sample equivalent)
25 ng/mL mixed working standard-in-matrix (50.0 ng/g sample equivalent).
- Pipet a portion of the standard mix into a 2 mL amber flat bottom autosampler vial for analysis.
- Working mixed standards-in-matrix of ciprofloxacin and enrofloxacin are good for the analysis of samples within a 24 hour period of time from initial standard preparation. See note 5.7.7.
- Note: Enrofloxacin and ciprofloxacin can exhibit breakdown under certain environmental conditions. To minimize this occurrence working mixed Standards are prepared at the time of analysis from vials of refrigerated stock solutions. Standards and samples are placed in amber vials or stored in low light conditions to minimize any photo-degradation of the antibiotics. Once a stock standard solution vial is removed for use in making the working mixed standard, the unused portion should be discarded and a new vial used for any additional standard preparation. Working mixed standards of ciprofloxacin and enrofloxacin are good for the analysis of samples within a 24 hour period of time from initial standard preparation. Ciprofloxacin is a major metabolite of enrofloxacin. The ratio of the two compounds in the working mixed standard can be monitored during the analytical run as an additional precaution against standard degradation during the analysis.
- 5.8 QC Blanks and Spikes
- Reagent blanks are run with each batch and are prepared by taking 7.5 mL of deionized water and running through the procedure along with the honey samples.
- Instrument blanks are prepared by placing a portion of the 16/84% acetonitrile/acetic acid (2%) solution used for sample reconstitution in an amber autosampler vial for instrumental analysis.
- Method spikes at 5.0 ng/g (action level) are prepared by taking 125 µL each of ciprofloxacin and enrofloxacin 0.1 ng/ µL stock standards and adding these to 2.5 grams of blank honey (± 0.03 grams) prior to extraction.
- Additional spikes at levels other than 5.0 ng/g may be necessary depending on the levels found in samples. Similar volumetric amounts of the standards should be utilized to minimize any effects on the extraction.
- Acceptance criteria for method spikes recoveries should be between 60-110% with a 20% CV. (Initial single laboratory validation and day-to-day variability in a single Tupelo honey showed an average recovery and process sigma of 72 ± 6 % for ciprofloxacin and 89 ± 6% for enrofloxacin). See Attachment F for summary of typical recoveries based on initial method validation and day to day spike recoveries versus external standards in tupelo honey.
- 5.9 Identification, Confirmation, and Quantification
- Fluoroquinolone identification requires at least three structurally significant ions. For each analyte, two ions are isolated and subsequently fragmented, resulting in at least two explicit ions per compound and one ion ratio. See section 5.5.7.
- The ion ratio must be within ± 10% of the average of the reference standards in a given analytical run as calculated by addition and subtraction. For example, at 50% relative abundance, the acceptability range would be 40-60%, not 45-55%. Signal to noise should exceed 3:1. Standards which fail to meet method performance criteria such as consistent instrument response are not included in this average. In cases where the sample concentration varies significantly from the standard, ion ratios are compared to standards at concentrations close to that of the sample. Preliminary analyses often show comparisons to a single standard and are confirmed using the average of the reference standards in a re-extraction and analysis run.
- The retention time of the sample peak must match the average reference standard peaks within +/- 0.25 minutes.
- 5.10 Reporting
- Samples below 2.5 ng/g will be reported as ND (no detect).
- Samples at or above 2.5 ng/g but less than 5.0 ng/g will be reported as BQL (below quantification limit)
- Sample at or above 5.0 ng/g will be reported as a numerical value unless otherwise noted in the batch comments.
-
-
Attachments
Attachment A: Instrument Method
Attachment B: Example of a Tune File
Attachment C: Example of an Extracted Ion Chromatogram
Attachment D: Example of an Analytical Run Sequence
Attachment E: Quantification using Standard Additions
Attachment F: Typical Method Recoveries versus External Standards in Tupelo Honey - References
- Hammack, W., et.al., Preparation and LC/MS/MS Analysis of Fluoroquinolones in Honey, Florida Department of Agriculture and Consumer Services Method CR 405, Revision 4, September, 1 2006.
- "Concurrent Determination of Four Fluoroquinolones; Ciprofloxacin, Enrofloxacin, Sarafloxacin and Difloxacin in Atlantic Salmon Tissue by LC with Fluorescence Detection," Jose E. Roybal, Calvin C. Walker, Allen P. Pfenning, Sherri B. Turnipseed, Steve A. Gonzales, and Jeffery A. Hurlbut, FDA Animal Drugs Research Center, Denver, CO, Oct 24, 2003, unpublished method.
- J AOAC Int. 2002 Nov-Dec; 85(6):1293-1301, Roybal, J, et.al., Concurrent Determination of Four Fluoroquinolones in Catfish, Shrimp, And Salmon By Liquid Chromatography With Fluorescence Detection.
- FDA LIB 4108, Turnipseed, S, et.al., Confirmation of Fluoroquinolones in Catfish Tissue by Electrospray LC/MS
- FDA LIB 4298, Turnipseed, S, et.al., Confirmation of Fluoroquinolone Residues in Salmon and Shrimp Tissue by LC/MS: Evaluation of Single Quadrupole and Ion Trap Instruments
- JAOAC Int., 2005; 88(4), pp 1160-1166, Schneider, M, et.al., Multiresidue Determination of Fluoroquinolones in Shrimp by Liquid Chromatography-Fluorescence-Mass Spectrometry
- J Chromatography A, 2002; 982(1), pp 97-109, Johnston, L, et.al., Determination of Quinolones and Fluoroquinolones in Fish Tissue and Seafood by High-Performance Liquid Chromatography with Electrospray Ionization Tandem Mass Spectrometric Detection
- REVISION HISTORY:
Rev Date Description Author 1 09-15-06 FDACS method CR 405 Revision 4 dated 09/01/06 formatted for FDA use. P. Kijak 2 09-29-06 Corrected limit in scope to 2.5 ppb, converted rpm to RCF P. Kijak
Attachment A: Instrument Method
Instrument Method: FLUROQUINOLONE_CE_HIFLOW.methAgilent1100 AutoSampler
Drawing speed (µL/min): 200
Ejecting speed (µL/min) : 200
Needle draw position offset (mm) : 0.0
Injection volume (µL) : 10.0
Wash vial: 100
Wash Cycle: 1
Wash Stroke: 20.0
Analysis stop time (min): 32.10
Post run time (min) : 0.00Timed events:
Time Contact Number Contact State 0.00(min) Agilent1100 Binary Pump
Solvent A: H2O/0.1% FORMIC
Solvent B: ACN/0.1% FORMIC
Minimum pressure limit (bar): 0.0
Maximum pressure limit (bar): 400.0
Post run time (min) : 0.00Gradient program:
Time Flow Rate Composition 0.00(min) 0.20(mL/min) A=95.0% B=5.0% 15.00(min) 0.20(mL/min) A=5.0% B=95.0% 25.00(min) 0.20(mL/min) A=5.0% B=95.0% 25.10 (min) 0.50 (mL/min) A=95.0% B=5.0% 30.00(min) 0.50(mL/min) A=95.0% B=5.0% 30.10(min) 0.20 (mL/min) A=95.0% B=5.0% 32.10(min) 0.20(mL/min) A=95.0% B=5.0% Agilent1100 Heater
Oven: On
Separate Mode: On
Post run time (min): 0:00Time Left Temperature Right Temperature Valve Position 0.00(min) 40.00(C) 40.00(C) 1 32.10(min) 40.00(C) 40.00(C) 1 LTQ Instrument Method
Creator: LTQ
Last modified: 9/1/2006 by LTQMS Run Time (min) : 32.10
Sequence override of method parameters not enabled.
Divert Valve: in use during run
Divert Time (min) Valve State 0.00 To Waste 5.05 To Source 15.05 To Waste Contact Closure: not used during run
Syringe Pump: not used during run
MS Detector Settings:
Additional Microscans:
MS2 0
MS3 0
MS4 0
MS5 0
MS6 0
MS7 0
MS8 0
MS9 0
MS10 0Segment 1 Information
Duration (min) : 32.l0
Number of Scan Events: 2
Tune Method: STATESCREEN_122804Scan Event Details:
1: ITMS + c norm !corona !pi · (332.0)->o(90.0-342.0)
MS/MS: CE 35.0% Q 0.250 Time 30.000 IsoW 3.0
2: ITMS + c norm !corona !pi · (360.0)->o(95.0-370.0)
MS/MS: CE 35.0% Q 0.250 Time 30.000 IsoW 3.0Custom Data Dependent Settings:
Not enabled
Attachment B: Example of a Tune File
510612E
STD HALFACTIONX_#1
Segment : 1Tune File Values Capillary Temp (C) : 300.00 APCI Vaporizer Temp (C) : 0.00 Sheath Gas Flow () : 50.00 Aux Gas Flow () : 10.00 Sweep Gas Flow () : 15.00 Source Type : ESI Injection Waveforms : Off Zoom AGC Target : 1000.00 Full AGC Target : 30000.00 SIM AGC Target : 10000.00 MSn AGC Target : 10000.00 POSITIVE POLARITY Source Voltage (kV) : 4.00 Source Current (µA) : 100.00 Capillary Voltage (V) : 9.00 Tube Lens (V) : 100.00 Skimmer Off set (V) : 0.00 Multipole RF Amplifier (Vp-p) : 400.00 Multipole 00 Offset (V) : -5.00 InterMultipole Lens 0 Voltage (V) : -4.50 Multipole 0 Offset (V) : -5.25 InterMultipole Lens 1 Voltage (V) : -10.00 Gate Lens Offset (V) : 0.00 Multipole 1 Offset (V) : -6.50 Front Lens (V) : -6.00 Zoom Micro Scans : 1 Zoom Max Ion Time (ms) : 50.00 Full Micro Scans : 1 Full Max Ion Time (ms) : 50.00 SIM Micro Scans: 1 SIM Max Ion Time (ms) : 50.00 MSn Micro Scans : 1 MSn Max Ion Time (ms) : 200.00
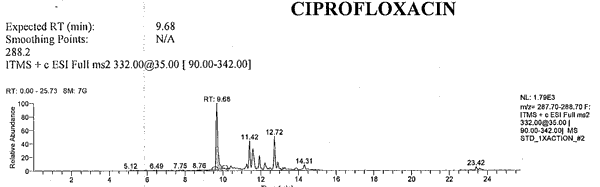
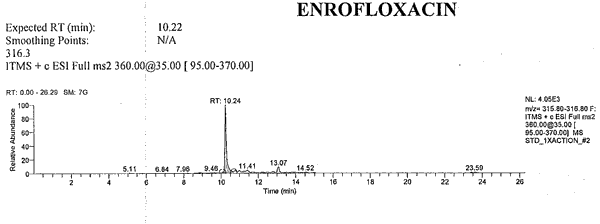
Attachment C: Example of an Extracted Ion Chromatogram
Data File Sample Name: STD_1XACTION_#2
Sample Type: Unknown
Acquistion Date: 08/15/06 22:48:35
Run Time (min): 32.10
Vial: 2
Injection Volume (µL): 10.00
Multiplier: 1.00
Comments: 510611E
Instrument Method: FLUROQUINOLONE_CE_HIFLOW.meth
Processing Method: None
Current Data Path: RUN#1_MSMSExpected RT RT Name Area Height Calculated Amount Specified Amount Units 9.68 9.68 CIPROFLOXACIN 11613 1762 5.212 N/A 10.22 10.24 ENROFLOXACIN 24312 3991 4.727 N/A
Attachment D: Example of an Analytical Run Sequence
Position File Name Inj Vol Sample Type Level Dil Factor 1 EQUIL01 10 Unknown 1 1 EQUIL02 10 Unknown 1 1 EQUIL03 10 Unknown 1 1 EQUIL04 10 Unknown 1 1 STD_2.5_#1 10 Std Bracket 2.5 PPB 1 2 STD_5_#1 10 Std Bracket 5 PPB 1 3 STD_10_#1 10 Std Bracket 10 PPB 1 4 STD_25_#1 10 Std Bracket 25 PPB 1 5 STD_50_#1 10 Std Bracket 50 PPB 1 6 Instrument Blank#1 10 Unknown 1 7 Control Honey Extract 10 Unknown 1 8 Control Honey Extract 10 Unknown 1 9 Fortified Honey Extract 10 Unknown 1 10 Fortified Honey Extract 10 Unknown 1 11 Sample 10 Unknown 1 12 Sample 10 Unknown 1 1 STD_2.5_#2 10 Std Bracket 2.5 PPB 1 2 STD_5_#2 10 Std Bracket 5 PPB 1 3 STD_10_#2 10 Std Bracket 10 PPB 1 4 STD_25_#2 10 Std Bracket 25 PPB 1 5 STD_50_#2 10 Std Bracket 50 PPB 1 6 Instrument Blank#2 10 Unknown 1 13 Sample 10 Unknown 1 14 Sample 10 Unknown 1 15 Sample 10 Unknown 1 16 Sample 10 Unknown 1 17 Sample 10 Unknown 1 18 Sample 10 Unknown 1 19 Sample 10 Unknown 1 20 Sample 10 Unknown 1 1 STD_2.5_#3 10 Unknown 1 2 STD_5_#3 10 Unknown 1 3 STD_10_#3 10 Unknown 1 4 STD_25_#3 10 Unknown 1 5 STD_50_#3 10 Unknown 1 Standard levels reflect concentration of target analyte in sample.
Action level = 5.0 ng/g
Attachment E: Quantification using Standard Additions
Standard additions
Quantification using standard additions may be necessary to improve accuracy in cases where the ion suppression or enhancement effects in a sample matrix are not adequately compensated for by the use of external standards prepared in the reference matrix. This technique utilizes known amounts of the target analyte added to the extracted sample so as to compensate for ion suppression or enhancement effects. Such effects have been observed when some other types of honey matrices were compared with the honey matrix used for the initial validation. Prior to standard additions, it will be necessary to make a rough estimate of the concentration of the target analyte in the sample using external standards as listed in sections 5.6.3. The procedure outlined in this attachment may then be utilized for the standard additions quantification. Variations of this procedure may also be necessary depending on the nature of the sample and the concentration of the target analyte. Adequate documentation of the standard additions experiment is vital.
One mL aliquots of sample extract are placed in each of three 2 mL size autosampler vials. These are labeled A, B, and C. Vials (B) and (C) are spiked with known amounts of the target analyte to produce signal counts roughly double (B vial), and triple (C vial) the counts present in the zero level sample (A vial). The volume of standard solution added to the (B) and (C) sample vials should be equal. Therefore, the use of standard solutions at the desired concentrations will be required. The same volume of blank solvent (solvent the spikes are prepared in) should be added to the (A) vial. Thus, A, B and C extracts are diluted to the same volume which simplifies the calculations necessary to obtain a final sample concentration. The target additions of standard to vials (B) and (C) are made based on estimates from the external standard quantification. These will only be estimates and may result in the signal counts in the (B) and (C) vials which do not provide reasonable detector response or regression and quantification. Additional estimates and repeated experiments may be necessary.
Once the sample extracts are spiked, each is analyzed and the area counts recorded for the target analyte in each of the A, B, and C vials. These values are then plotted vs the concentrations of the additions in each vial using a linear regression fit with the area counts plotted on the vertical axis (Y) and the concentrations of the additions plotted on the horizontal axis (X). Note: vial (A) has a concentration of (0) as no standard was added to this sample. The equation is then solved for (X) when (Y) equals zero. The absolute value of (X) is then corrected for the dilution resulting from the addition of the standard and solvent to vials A, B, and C. This corrected value is then multiplied by the concentration of sample by weight (grams solids analyzed) in the final extract (5.0 mL/2.5 gm) to obtain the final concentration of analyte in the sample. For example, if samples in vial A, B, and C are all diluted with 100 ul of standard or blank solvent then the correction value is (1.1/1.0) or 1.1 . This value would be multiplied by the value obtained from the curve and then by a factor of (5.0/2.5) or 2 to arrive at the final concentration of analyte in the sample in ng/g. If the extracts are diluted prior to the standard additions experiment this must also be factored into the final calculation.
Example of estimating the concentration of a target analyte in final extract from external standard quantification:
If a sample is calculated to contain a level of 5 ng/g of target analyte, then the concentration present in the final extract is as follows:
(5 ng/g) × (2.5 g) = 12.5 ng of target in final volume of 5 mL = 2.5 ng/mL in final extract.
To approximately double this concentration it is necessary to place 2.5 ng of target in the 1 mL aliquot of final extract used for the standard addition experiment. A standard with a concentration level of 0.05 ng/ul would require 50 ul of standard be pipeted into the 1 mL aliquot for an actual addition concentration of 2.5 ng/1.05 mL or 2.38 ng/mL. This would be sufficient to approximately double the concentration in the sample. The exact value of 2.38 would be plotted on the horizontal axis (X) vs the area counts obtained for this sample when measured by the analytical instrument.
Important points:
The fit of the linear regression curve should be greater than 0.99
All samples should be diluted to the same volume for simplification of the calculations.
Attachment F: Typical Method Recoveries versus External Standards in Tupelo Honey
Ciprofloxacin Fortification Level (ppb) Average Recovery (%) Standard Deviation CV (%) Number of Samples 2.5 69 2 3 3 5.0 74 7 9 21 10 66 11 16 5 100 68 5 7 5 Enrofloxacin Fortification Level (ppb) Average Recovery (%) Standard Deviation CV (%) Number of Samples 2.5 95 3 3 3 5.0 90 7 8 21 10 85 16 19 5 100 90 3 3 5
Chemical Name: ciprofloxacin
IUPAC International Chemical Identifier:Chemical Name: enrofloxacin
IUPAC International Chemical Identifier: