

GUIDANCE DOCUMENT
Prospective Manufacturers of Barrier Devices Used During Oral Sex for STD Protection October 1996
- Docket Number:
- FDA-2020-D-0957
- Issued by:
-
Guidance Issuing OfficeCenter for Devices and Radiological Health, Office of Product Evaluation and Quality
DEPARTMENT OF HEALTH & HUMAN SERVICES
Public Health Service
Food and Drug Administration
9200 Corporate Boulevard
Rockville MD 20850
DATE: October 31, 1996
TO: Prospective Manufacturers of Barrier Devices Used During Oral Sex for STD Protection
SUBJECT: Regulatory Requirements for Obtaining Marketing Clearance in the United States
This letter is to advise you of a change in the regulatory requirements for barrier devices intended for protection from sexually transmitted diseases (STDs) during oral sex. As you may know, the Food and Drug Administration (FDA) advised manufacturers in the past that barrier devices for STD protection during oral sex needed an approved premarket approval application (PMA) in order to place these devices into commercial distribution in the United States. FDA has reconsidered the regulatory requirements applicable to these devices to ensure consistency with the requirements for other barrier devices that are used to prevent disease transmission. In particular, FDA considered condoms, exam and surgical gloves, as well as surgical gowns, masks, and drapes. All of these Class I and Class II devices protect an anatomical site from transfer of microorganisms, body fluids, and particulate material. Latex condoms are the most widely recognized example of a barrier product to protect users from the transmission of STD microorganisms during sexual relations.
Barrier products provide protection from various routes of disease transmission. FDA recognizes that the safety and effectiveness of barrier devices for oral sex present the same issues as barriers for other applications, i.e., biocompatibility, barrier integrity, and device placement. It is for these reasons that we believe that barrier devices for oral sex can be properly evaluated through the premarket notification (510(k)) process, rather than through the PMA process.
Similar to 510(k)s for other devices used to prevent disease transmission, a 510(k) for a barrier device for oral sex should provide bench testing data to establish adequate performance under simulated use conditions. In particular, the 510(k) should contain data from in-vitro studies to demonstrate the barrier integrity with respect to STD microorganisms. Enclosed please find a test methodology developed by FDA for evaluating the barrier properties of condoms. While the method would require modification for barriers designed for oral sex, it may provide some insight into data that should be provided in a marketing application. In addition, please keep in mind that certain device designs may necessitate the collection of data to demonstrate that the user can safely and effectively place the device.
The Division of Reproductive, Abdominal, Ear, Nose and Throat, and Radiological Devices (DRAERD), Office of Device Evaluation, developed a package for preparing a 510(k). This package on 510(k) content and format, entitled "Explanation for Items in DRAERD Premarket Notification 510(k) Screening Checklist", is enclosed. In addition, the Division of Small Manufacturers Assistance (DSMA) can be contacted at (800) 638-2041 or (301) 443-6597 to request a copy of a manual entitled "Premarket Notification 510(k): Regulatory Requirements for Medical Devices."
You may not market such a barrier device for oral sex use until you have filed a 510(k) in accordance with 21 CFR 807.87, and you have received a letter from FDA authorizing you to do so. If you market such a device without conforming to these requirements, you will be in violation of the Federal Food, Drug, and Cosmetic Act (Act). You may, however, distribute such a device for investigational use to obtain clinical information to establish substantial equivalence. Depending on the study and device design, an investigation may be conducted under the abbreviated investigational device exemption (IDE) requirements (21 CFR 812.12(b)).
If you are developing, or plan to develop, a device intended for STD protection during oral sex, you are strongly encouraged to contact FDA's Obstetrics and Gynecologic Devices Branch to discuss the type of testing needed to support a 510(k). If you have any questions, please contact Mr. Colin M. Pollard, Chief, Obstetrics and Gynecology Devices Branch, at (301) 594-1180.
Sincerely yours,
Philip J. Phillips
Deputy Director
Office of Device Evaluation
Center for Devices and Radiological Health
Enclosures
- FDA Guide to Determining Barrier Properties of Condoms to Virus Penetration
- Explanation for Items in DRAERD Premarket Notification 510(k) Screening Checklist
From: CDRH Testing Guidance for Male Condoms Made from New Materials, issued June 29, 1995.
ATTACHMENT A- INFORMATION FOR DETERMINING BARRIER PROPERTIES OF CONDOMS TO VIRUS PENETRATION
OFFICE OF SCIENCE AND TECHNOLOGY
CENTER FOR DEVICES AND RADIOLOGICAL HEALTH
Introduction
This guideline addresses the rationale, methodology and required sensitivity of a test of the ability of condoms to act as barriers to transmission of the etiological agents of sexually-transmitted diseases (STDs), including viruses. The condom is identified in the code of federal regulations (CFR) at Title 21 CFR Section 884.5300 as ..."a sheath which completely covers the penis with a closely fitting membrane. The condom is used for contraception and prophylactic purposes (preventing transmission of venereal disease)." The device may also be used to collect semen to aid in the diagnosis of infertility.
A medical claim for condoms as being effective against STDs requires that appropriate laboratory tests be performed. Since viruses are the smallest etiological STD agents and include the human immunodeficiency virus (HIV) and hepatitis B virus (HBV), the challenge particle should be a small virus or virus-size particle. Test conditions should account for as many parameters as possible that are considered to be important in real-life conditions. Appropriate choices of challenge particle, solution properties, and test pressure and duration are considered most important and must be included. The barrier properties of a condom may be determined in a static test, i.e., movement of the condom during the test is not required. Choices of parameters that make the in vitro test more stringent than expected real-life use are encouraged, with appropriate justification.
The choice of challenge particle has several important aspects. A biological assay may be preferred in general because there should be no "background" level of confounding "signal," as would be found with radioactively- or otherwise-labeled viruses or virus-like particles. Surrogate viruses of appropriate size and shape may substitute for human pathogens. Such surrogates may be bacterial viruses (bacteriophages), which are safer, faster and less expensive to use for testing and which can be readily obtained at sufficient titer to provide an adequate challenge concentration. However, in order for the test to be used to demonstrate safety with regard to STDs, the test virus should be smaller than hepatitis B virus (42 nm diameter), the smallest etiological agent for a STD. For these reasons, the following protocol suggests use of a small bacterial virus as challenge particle.
Preparation of Test Samples
Test condoms should be carefully handled so they are not damaged during the test procedure. Gloves may be worn as a precautionary measure to prevent abrasion or puncture by fingernails, rings, etc.
Most of accompanying lubricants and/or spermicides, if present, should be removed so they don't interfere with the test. They may be removed by rinsing with buffer and gently patting dry.
The Basic Test
The test consists of filling the condom with virus-containing buffer and determining whether any viruses penetrate that barrier during submersion in collection buffer. Virus penetration is quantitated and reported as equivalent volume of challenge suspension needed to account for amount of virus penetration. The basic methodology using simple, readily-available equipment has been published (see Lytle et al reference). More sophisticated apparatus (see Retta et al reference) may be used to make the testing more convenient, although the basic test parameters should remain similar. The elements of the test should include:
- attaching the test condom to an apparatus which:
- provides a leakproof seal around the top and leaves an appropriate length of test portion available for the virus penetration test (at least 140 mm);
- provides for restraining of the condom to prevent overexpansion under pressure (Dimensions of the restrainer should allow expansion of the test portion of the condom to a length of 140-150 mm and a circumference of 120-130 mm. The contour of the restrainer should match that of the condom, including the reservoir tip, if present. Restrainers of the same size and material should be used with the test condoms and with the comparative condoms. In the case of a condom that is larger or smaller than a standard, an appropriate size restrainer should be used to accommodate the dimensions of the condom (must be justified).
- provides for exposure of the inside of the condom to aqueous challenge virus suspension;
- provides for application of pressure to that suspension;
- allows for submersion of test portion of condom in collection fluid; and
- provides for access to challenge virus suspension inside condom for assay following the test.
- filling the condom with a buffer that:
- has appropriate properties (pH approximately 7.0, salinity of any one of several variations of physiological saline, surface tension less than 0.05 N/m [may be provided by 0.1% Triton X-100]) (Physiological saline has a lower viscosity than semen and therefore provides a more stringent test. The test may be performed at room temperature [68-72 øF] when saline is used); and
- contains the challenge virus at sufficient titer, even at the end of the test (at least 108 plaque forming units/mL of a small, approximately spherical virus). The bacteriophage ØX174 may be used as the challenge virus. In the case of a virus other than ØX174, its use must be justified.
- providing pressure to the challenge fluid equivalent to 60 mmHg (1.28 psi) or more (e.g., hydrostatically with a 810 mm column of water or with air/gas pressure);
- providing a collection container with sufficient buffer to allow fluid contact with the test surface of the condom and to collect any virus that penetrates through the condom;
- submerging the filled, pressurized condom (first 140 mm from the closed end, not including the reservoir tip, if present) in the collection buffer for at least 30 minutes;
- assaying the collection buffer for the challenge virus to determine whether any virus has penetrated the condom and passed into the collection buffer (The collection fluid must be mixed at the time of assay so that the assay aliquots are representative.); and
- calculating the equivalent volume of challenge virus penetration needed to account for amount of virus found in collection buffer.
Controls
It is known that some viruses can be removed from suspension by certain materials through binding, or that they can be rendered biologically undetectable by chemical inactivation. Thus controls are needed to assure that the virus penetration test will yield meaningful data. Positive control experiments of the same duration are needed to assure that the overall test is functioning properly. Condoms with intentional pinholes may be used, although it is recognized that it is difficult to produce small pinholes.
In addition, it must be ascertained whether the challenge virus remains at a stable concentration in the condom during the test. Data from several condoms are needed and must be collected as part of each condom test. The titer of the challenge virus suspension inside the condom at the end of the test is compared to the titer originally placed in the condom. This determines if and how much the challenge virus titer changes during the test because of interaction with the condom and the test apparatus, or other factors.
It must also be ascertained whether any virus that penetrates the condom remains detectable in the collection buffer over the test period. This can be done by "spiking" the collection buffer with a low level of virus before a mock test (where there is no virus inside the condom and for the same duration) and assaying the titer of the collection buffer at the beginning and end of the mock test. This determines if and how much the penetrated virus titer changes during the test as a result of interaction with the outside of the condom, the restrainer or the collection container.
If either (or both) of the above controls indicates loss of virus titer, the starting challenge titer must be increased to compensate for the loss in order to maintain the overall sensitivity of the test.
It may be useful to determine via controls (e.g., settle plates) whether contamination caused by aerosolized virus or other leaks might lead to false evidence of virus penetration of the condom.
Sampling Procedure
A complete data set should include results from at least 60 condoms (20 condoms from each of 3 lots), in order to provide assurance that overall quality of each of three lots is satisfactory.
Comparative (predicate) samples
Latex condoms (off-the-shelf) are to be used. However, since the history (duration and temperature of storage) of such samples is not known and may affect the integrity of the samples, these samples must be used before the expiration date and should give virus transmission rates similar to those reported in the published literature. We suggest using non-lubricated, smooth (not ribbed, non-reservoir tip) samples. They should be treated in the identical manner as the investigational test samples, including mock removal of lubricant/spermicide, if appropriate.
Detection Limit
A typical method to determine the virus titer in the collection buffer would be to assay 1 mL in triplicate (3 mL total). In order to have 95% confidence that an assay will find at least one virus when virus is present [i.e., P(0) <0.05], the="" average="" number="" of="" infectious="" particles="" per="" total="" volume="" assayed="" must="" be="" at="" least="" three;="" e.g.,="" there="" is="" a="" 95%="" probability="" that="" a="" titer="" of="" 1="" pfu/ml="" will="" result="" in="" at="" least="" one="" plaque="" in="" a="" 3="" ml="" total="" assay.="" thus,="" the="" sensitivity="" or="" detection="" limit="" of="" this="" assay="" can="" be="" claimed="" as="" 1="" pfu/ml="" when="" 3="" ml="" is="">
Detection limit expressed as volume of challenge virus suspension that penetrated the barrier is probably the most useful measure of test sensitivity. For example, in a real-life risk assessment the volume of transmitted virus-containing fluid can be translated into infectious units when the titer of a pathogenic virus (in real life) is known.
The test procedure must be able to detect 2 x 10-6 mL penetration of the challenge virus suspension. This can be done by using a challenge titer of 1 x 108 pfu/mL, a collection buffer volume of 200 mL and assaying 1 mL in triplicate from the collection buffer (assuming no loss of virus titer in the challenge buffer nor in the collection buffer): the assay detection limit of 1 pfu/mL is equivalent to penetration by 200 pfu (1 pfu/mL x 200 mL) or 2 x 10-6 mL (200 pfu divided by 1 x 108 pfu/mL).
Presentation of Results
A table of the results for all the test condoms should be presented that includes: the challenge virus titer, the virus titer in the collection buffer, any correction factor for loss of virus (determined in the controls), and the calculated challenge volume that penetrated (for the condoms that allowed virus transmission). (See example below, Table I.) The volume of challenge virus suspension needed to account for the virus penetration into the collection buffer can be calculated for each condom by the method presented in the previous section.
If some loss of virus titer occurs either inside the condom or outside in the collection container, the calculation should include the appropriate correction for such loss. For condoms that apparently did not allow virus transmission, the detection limit of that particular test should be given, e.g., as 2 x 10-6 mL.
Report Forms
Test results for virus penetration of condom samples should be presented in tabular form, where the data for each condom are individually reported. Necessary items for each test sample are:
- date test was performed,
- titer of challenge titer inside the condom at the end of the test,
- calculated detection limit based on the challenge virus titer, the collection fluid volume, and the volume assayed,
- pfu's found in aerosol control,
- pfu's detected in the collection fluid,
- calculated titer of penetrated virus in collection fluid, and
- volume of challenge virus suspension needed to account for the amount of virus detected in the collection fluid.
Information accompanying the table should include:
- the challenge virus,
- the challenge and collection fluids (e.g., buffer and surfactant),
- how the titer of the challenge virus suspension was determined (dilution, volume assayed, and number of replicate assays),
- the challenge volume (if variable from one test sample to another, it should be included in the table for each condom),
- the collection fluid volume (if variable from one test sample to another, it should be included in the table for each condom),
- the transmembrane condom pressure and how it was provided (if variable from one test sample to another, it should be included in the table for each condom), and
- any evidence of an equipment or procedural malfunction during any particular test.
Table I. Results for virus penetration through condom samples of Brand X, Lot #34068.
| i | ii Titer challenge virus |
iii Detect limit |
iv Aerosol control |
v Collect buffer |
vi Titer, collect buffer |
vii Volume penetra virus (mL) |
|
|---|---|---|---|---|---|---|---|
| 1 | 10/28 |
2.2x108 ±0.2) |
0.9x10-6 |
0,0,0 |
29,28,33 |
3.0x101 (±0.2) |
2.7x10-5 |
| 2 |
10/28 |
2.3x108 (±0.2) |
0.9x10-6 |
0,0,0 |
0,0,0 |
<> | <>-6 |
Positive Control
Reporting the results of the positive control experiment should be done using the same reporting format as with virus penetration of test samples.
Control to Test Challenge Virus Stability
Results from the test of challenge virus stability should be presented in tabular form, where the data for each condom are individually reported. (See example below, Table II.) Necessary items for each test sample are:
- date test was performed,
- titer of challenge titer placed inside the condom at the beginning of the test,
- titer of challenge titer inside the condom at the end of the test, and
- calculated ratio of final to beginning titer.
Table II. Results of test for stability of challenge virus in condom samples of Brand X, Lot #34068.
| Sample | Date | Beginning titer (pfu/mL) |
Final titer (pfu/mL) |
Ratio, final/ begin |
|---|---|---|---|---|
| 1 | 10/28/93 | 2.2x108 (±0.2) |
2.1x108 (±0.2) |
0.95 |
| 2 | ||||
| 3 |
Control to Test Detection of Virus Which Penetrates Condom ("Spiking experiment")
Results from tests to determine the detection of penetrated virus should be in tabular form, where the data for each condom are individually reported. (See example below, Table II.) Necessary items for each test sample are:
- date test was performed,
- virus titer in collection buffer at the beginning of the test,
- virus titer in collection buffer at the end of the test, and
- calculated ratio of final to beginning titer.
Table III. Results of test for detection of penetrated virus in contact with condom samples of Brand X, Lot #34068.
| Sample | Date | Beginning titer (pfu/mL) |
Final titer (pfu/mL) |
Ratio final/ begin |
|---|---|---|---|---|
| 1 | 10/28/93 | 1.2x102 (±0.1) |
1.1x102 (±0.1) |
0.92 |
| 2 | ||||
| 3 |
References
Lytle, Routson and Cyr. A simple method to test condoms for penetration by viruses. Appl. Environ. Microbiol. 58: 3180-3182, 1992.
Retta, Herman, Rinaldi, Carey, Herman and Athey. Test method for evaluating the permeability of intact prophylactics to viral-size microspheres under simulated physiologic conditions. Sex. Trans. Diseases 18: 111-118, 1991.
RRG/LLD 1/6/93 DRAERD Premarket Notification 510(k)
Rev. 2/6/96 Screening Checklist
510(k) Number & Device Name
Company
MORE
ITEM PRESENT NEEDED
Yes No (Y/N/?)
1. General information (i.e., trade & classification name,
Est. Reg. No., device class, meets special
controls or performance standards, etc.)
Reason for 510(k) - new device or modification
Identification of legally marketed equivalent device
Truthful and accurate statement
SMDA 510(k) summary
2. Proposed Labeling, Labels, Advertisements
Description of new device/modification
Intended use statement
Diagrams, Engineering Drawings, Photographs
Indication for Use Statement
3. Comparison of similarities/differences to named
legally marketed equivalent device
Equivalent Device Labeling, Labels, Advertising
Intended use of equivalent device
4. List of all patient contacting materials in new device
Comparison of materials to equivalent device
5. Biocompatibility information/data for patient
contacting materials, OR
Certification - identical material/formulation
6. Performance data: Bench data
Animal data
Clinical data
7. Sterilization information
8. Software validation & verification
9. If Class III, Class III Certification & Summary
10. If kit, kit certification
RRG Rev. 2/6/96
for Items in
DRAERD Premarket Notification 510(k)
Screening Checklist
The Division of Reproductive, Abdominal, Ear, Nose and Throat, and Radiological Devices (DRAERD) of the Office of Device Evaluation (ODE) has initiated a screening program for all 510(k) premarket notifications received in the division. The purpose of this screening program is to provide the submitter of a 510(k) immediate feedback on basic administrative completeness of the initial submission before we begin our technical review. In the past, a majority of the submissions received in DRAERD were found to be lacking in basic information necessitating requests for additional information. These requests for additional basic information have resulted in delays for both the 510(k) submitter and the Food and Drug Administration (FDA).
Below is an explanation of each item on the screening checklist. There are three (3) columns of "blanks" with the first or second column having a "check mark," and the third having a "Y" (for yes), a "N" (for no), or a "?" (for don't know or unknown). A person submitting a 510(k) should look at the "Needed (Y/N/?)" column to see what additional basic information needs to be submitted. In addition, a specific item may be circled if only that item is missing. If the third column indicates a "?" (mainly applicable for item number 6, Performance data), we can not at this time determine if this information is needed. If you already have any information on a "?" item, it is suggested that you submit it as it will make the 510(k) more complete and may eliminate a later request for additional information. There may also be comments in the margin on the right side of the checklist. Checklist items:
1. General information includes the following:
a. applicant's name, signature and date, address, contact person and telephone number;
b. a table of contents, listing of tabs and appendices, and appropriate pagination;
c. the device name (trade or proprietary name and the common or usual name);
d. the classification name;
e. the establishment registration number, if applicable, of the owner or operator submitting the premarket notification;
f. the address of the manufacturing facility/facilities and, if appropriate, the sterilization site(s);
g. the class in which the device has been placed under section 513 of the act, and, its appropriate panel, if known, or, if the submitter determines that the device has not been classified, a statement of that determination and the basis for that determination;
h. action taken by the submitter to comply with the requirements of the Federal Food, Drug, and Cosmetic Act (act) under section 514 performance standards or section 513 special controls;
I. the reason for the premarket notification - a new device or a modification to an existing device (if modification, provide the 510(k) number for that device, if applicable);
j. an identification of the legally marketed device to which you claim equivalence. If known, provide the equivalent device's 510(k) number; and,
k. a statement that the submitter believes, to the best of his knowledge, that all data and information submitted in the premarket notification are truthful and accurate and that no material fact has been omitted.
l. The Safe Medical Devices Act of 1990 (SMDA) requires all persons submitting a premarket notification submission to include either (1) a summary of the safety and effectiveness information in the premarket notification submission upon which an equivalence determination could be based (510(k) summary), or (2) a statement that safety and effectiveness information will be made available to interested persons upon request (510(k) statement).
The content and format of a 510(k) summary can be found in Section 21 CFR 807.92. This Section states the following:
(a) A 510(k) summary shall be in sufficient detail to provide an understanding of the basis for a determination of substantial equivalence. FDA will accept summaries as well as amendments thereto until such time as FDA issues a determination of substantial equivalence. All 510(k) summaries shall contain the following information:
(1) The submitter's name, address, telephone number, a contact person, and the date the summary was prepared;
(2) The name of the device, including the trade or proprietary name if applicable, the common or usual name, and the classification name, if known;
(3) An identification of the legally marketed device that the submitter claims equivalence. A legally marketed device to which a new devices may be compared for a determination of substantial equivalence is a device that was legally marketed prior to May 28, 1976, or a device which has been reclassified from class III to class II or I (the predicate), or a device which has been found to be substantially equivalent through the 510(k) premarket notification process;
(4) A description of the device that is the subject of the premarket notification submission, such as might be found in the labeling or promotional material for the device, including an explanation of how the device functions, the scientific concepts that form the basis for the device, and the significant physical characteristics of the device, such as device design, material used, and physical properties;
(5) A statement of the intended use of the device that is subject of the premarket notification submission, including a general description of the diseases or conditions that the device will diagnose, treat, prevent, cure, or mitigate, including a description, where appropriate, of the patient population for which the device is intended. If the indication statements are different from those of the legally marketed device identified in paragraph (a)(3) of this section, the 510(k) summary shall contain an explanation as to why the differences are not critical to the intended therapeutic, diagnostic, prosthetic, or surgical use of the device, and why the differences do not affect the safety and effectiveness of the device when used as labeled; and
(6) If the device has the same technological characteristics (i.e., design, material, chemical composition, energy source) as the predicate device identified in paragraph (a)(3) of this section, a summary of the technological characteristics of the new device in comparison to those of the predicate device. If the device has different characteristics from the predicate device, a summary of how the technological characteristics of the device compare to a legally marketed device identified in paragraph (a)(3) of this section.
(b) 510(k) summaries for those premarket submissions in which a determination of substantial equivalence is also based on an assessment of performance data shall contain the following information:
(1) A brief discussion of the nonclinical tests submitted, referenced or relied on in the premarket notification submission for a determination of substantial equivalence;
(2) A brief discussion of the clinical tests submitted, referenced, or relied on in the premarket notification submission for a determination of substantial equivalence. This discussion shall include, where applicable, a description of the subjects upon whom the device was tested, a discussion of the safety and effectiveness data obtained from the testing, with specific reference to adverse effects and complications, and any other information from the clinical testing relevant to a determination of substantial equivalence; and
(3) The conclusions drawn from the nonclinical and clinical tests that demonstrate the device is as safe, as effective, and performs as well as or better than the legally marketed device identified in paragraph (a)(3) of this section.
(c) The summary should be in a separate section of the submission, beginning on a new page and ending on a page not shared with any other section of the premarket notification submission, and should ne clearly identified as a "510(k) summary."
(d) Any other information reasonably deemed necessary by the agency.
The content and format of a 510(k) statement can be found in Section 21 CFR 807.93. This Section states the following:
(a)(1) A 510(k) statement submitted as part of a premarket notification shall state as follows:
I certify that, in my capacity as (the position held in company by person required to submit the premarket notification, preferably the official correspondent in the firm), of (company name), I will make available all information included in this premarket notification on the safety and effectiveness within 30 days of request by any person if the device described in the premarket notification submission is determined to be substantially equivalent. The information I agree to make available will be a duplicate of the premarket notification submission, including any adverse safety and effectiveness information, but excluding all patient identifiers, and trade secret and confidential commercial information, as defined in 21 CFR 20.61.
(2) The statement in paragraph (a)(1) of this section should be signed by the certifier, made on a separate page of the premarket notification submission, and clearly identified as "510(k) statement."
(b) All requests for information included in paragraph (a) of this section shall be made in writing to the certifier, whose name will be published by FDA on a list of premarket notification submissions for which substantial equivalence determinations have been made.
(c) The information provided to requestors will be a duplicate of the premarket notification submission, including any adverse safety and effectiveness information, but excluding all patient identifiers, and trade secret and confidential commercial information, as defined in 21 CFR 20.61 of this chapter.
2. Proposed labels, labeling, advertising and/or promotional materials, and specifications sufficient to describe the new device/modification, its intended use, and directions for use, as appropriate. The label of most device packaging must bear the caution statement as outlined in 21 CFR 801.109 (b)(1): "CAUTION: Federal law restricts this device to sale by or on the order of a physician." Guidance on labeling issues is provided in ODE Bluebook Memo G91-1, "Device Labeling Guidance" dated March 8, 1991. To obtain a copy of this guidance, see below. Service manuals, engineering diagrams, drawings and/or photographs are usually necessary. Manufacturing information may be necessary.
On January 1, 1996, FDA began requiring that all 510(k) submitters provide on a separate page and clearly marked "Indication For Use" the indication for use of their device. (See attached Optional Format 1-2-96.) This indication for use statement should be an exact copy of the indication for use section in your device labeling.
3. A comparison table and discussion indicating how the device is similar to and different from other products of comparable type, that are legally marketed in the United States, accompanied by data to support the statement. Legally marketed devices are those device(s) on the market before May 28, 1976, (pre-amendments), or devices found "substantially equivalent" after 1976 through the 510(k) process. If this 510(k) is for a modification to a device, describe the modifications. It is helpful and time saving to re-supply information from the original submission (i.e. drawing, specifications, etc.) rather than just supplying the reference 510(k) number for FDA to look up the information. This information should include an identification of the substantially equivalent device(s), its intended use, its specifications, labels, labeling and advertising and/or promotional materials.
4. For patient contacting devices, a list of all materials used in the new/modified device with a comparison of these materials to the device to which you claim equivalence. You must specifically identify the materials; indicating "silicone" or "polyvinyl chloride" is not sufficient. Formulation information should be supplied for all polymers.
5. For patient contacting devices, biocompatibility information and/or data on the device materials. See the Tripartite Biocompatibility Guidance document for the appropriate testing to satisfy this requirement. In general, all testing should be on sterilized final devices. Alternatively, we may accept a certification from you that the exact same material and formulation is used in this new/modified device as was used in the device to which you claim equivalence.
6. Some devices and device modifications require performance data in order to determine equivalence. Performance data should be compared to the performance of the device to which you claim equivalency. Performance data includes all bench, animal, and clinical data collected with the new device/modification. Performance data include objectives, test set-up, protocol, results, discussion of results, appropriately supporting device specifications, and conclusions.
7. Sterility information, if labeled as sterile, includes the method of sterilization; the sterility assurance level (SAL); the method used to validated the sterilization cycle; if ethylene oxide sterilization is used, the residues levels for ethylene oxide (EtO), ethylene chlorohydrin (EtCh), and ethylene glycol (EtG); if radiation sterilization is used, the dose delivered; a description of the packaging, including materials; and if labeled as non-pyrogenic, provide the method used to make that determination and provide the sensitivity of the pyrogen assay used.
8. A report on software/firmware requirements, development, validation and verification is needed for all computer controlled devices dependent on software. A software release number should be indicated. Hardware validation and verification may also be required. The appropriate testing information should be submitted. This information is contained in Reviewer Guidance for Computer Controlled Medical Devices Undergoing 510(k) Review.
9. The SMDA also requires that any person who asserts that a device is substantially equivalent to a class III device to (1) certify that he/she has conducted a reasonable search of all information known, or otherwise available, about the generic type of device, and (2) provide a summary description of the types of safety and effectiveness problems associated with the type of device and a citation to the literature, or other sources of information, upon which they have based the description (class III summary and certification).
If claiming substantial equivalence to a Class III device, provide the following as described under Section 513(f)(3) of the act:
"I certify that, in my capacity as [The Position Held In Company], of [Company Name], I have conducted a reasonable search of all information known or otherwise available about the types and causes of safety or effectiveness problems that have been reported for the [Type Of Device]. I further certify that I am aware of the types of problems to which the [Type Of Device] is susceptible and that, to the best of my knowledge, the following summary of the types and causes of safety or effectiveness problems about the [Type Of Device] is complete and accurate:
[NUMBER AND LIST SEPARATELY EACH TYPE OF PROBLEM AND ITS CAUSES]
Attached is a bibliography, or other citation, of the materials upon which the above summary is based."
Printed name of person required to submit 510(k):
Signature of person required to submit 510(k):
Title of person submitting 510(k):
Name of Company:
Date:
10. If this 510(k) is for a kit, a certification or other information is required (see attachment).
Copies of all guidance documents may be obtained from the Division of Small Manufacturers Assistance, CDRH, FDA, at 800-638-2041 or 301-443-6597.
Attachments
Page of 510(k) Number (if known): Device Name: Indications for Use: (PLEASE DO NOT WRITE BELOW THIS LINE - CONTINUE ON ANOTHER PAGE IF NEEDED)
Concurrence of CDRH, Office of Device Evaluation (ODE) Prescription Use: OR Over-the Counter Use: (Per 21 CFR 801.109) (Optional Format 1-2-96)
3/14/95
For review purposes of a premarket notification (510(k)) for a kit, please provide the certification stated below:
I certify that the following components of my kit are either (1) legally marketed pre-amendments devices, (2) exempt from premarket notification (consistent with the exemption criteria described in the classification regulation(s) and the limitations of exemptions from Section 510(k) of the act (e.g., 862.9), or (3) have been found to be substantially equivalent through the premarket notification process for the use(s) for which the kit is to be intended (i.e., I am not claiming or causing a new use for the component(s)).
I further certify that these components are not purchased in "bulk", but are purchased in finished form, i.e., they are packaged, labeled, etc., consistent with their pre-amendments, exemption, or premarket notification criteria and status.
If you cannot make the above referenced certification statement (first paragraph) for each component of your kit, you must itemize the components without a pre-amendments, exemption, or premarket notification status. In this case we will continue our premarket notification review of these components of your kit.
If you cannot make the above referenced certification statement (second paragraph) for each component of your kit, you must itemize these components, state whether they are pre-amendments, exempt, or have been found substantially equivalent through the premarket notification process, and describe how you further process them (e.g., sterile, package/repackage, label/relabel, etc.).
If your kit contains examination gloves which are purchased in bulk, your submission must contain the following:
a. data demonstrating or certification that the final finished sterile examination gloves in the kit meet the American Society for Testing and Materials (ASTM) standards for rubber examination gloves, ASTM D 3578-77 (Reapproved 1991); and,
b. data demonstrating or certification that the final finished sterile examination gloves pass the FDA 1000 milliliter water leak test in accordance with the sample plan and test method published in the FEDERAL REGISTER (55 FR 51254-51258).
If the device kit contains components which are subject to regulation as drugs, a substantially equivalent determination will not apply to the drug component(s) of the device. For information on applicable Agency requirements for marketing the drug component(s) in the kit, it is suggested that you contact the Center for Drug Evaluation and Research, Division of Drug Labeling Compliance, at (301) 594-0063. Correspondence should be addressed as follows:
Center for Drug Evaluation and Research
Food and Drug Administration
5600 Fishers Lane
Rockville, Maryland 20857
If the kit contains sutures, provide evidence that the sterilant does not come into contact with the sutures during sterilization of the kit. Based on the evidence submitted, FDA will conclude if the sutures are or are not further processed. Inclusion of the sutures as components in your kit requires you to comply with the following conditions:
a. The labeling, packaging, and method of sterilization of the sutures you have listed cannot be changed without prior notification, review, and approval by FDA.
b. The suppliers of the sutures used in your kit cannot be changed without prior notification, review, and approval by FDA.
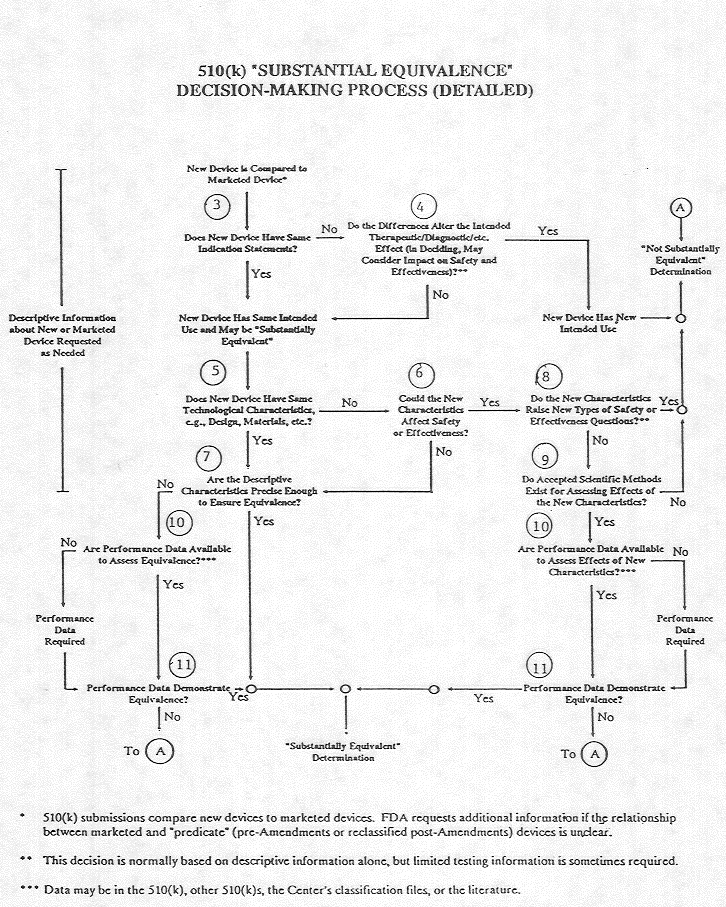
A 510(k) Substantial Equivalence Decision Making Process Flow Chart is available in pdf version or found directly below.
Submit Comments
You can submit online or written comments on any guidance at any time (see 21 CFR 10.115(g)(5))
If unable to submit comments online, please mail written comments to:
Dockets Management
Food and Drug Administration
5630 Fishers Lane, Rm 1061
Rockville, MD 20852
All written comments should be identified with this document's docket number: FDA-2020-D-0957.