

Statistical Review and Evaluation - Rotarix
Statistical Review and Evaluation
BLA#: 125265/0
Product: Rotarix, Live Attenuated Human Rotavirus (HRV) Vaccine, Oral (GSK)
Indication: Prevention of rotavirus gastroenteritis in infants and children caused by the serotypes G1, and non-G1 types (including G2, G3, G4, and G9).
Applicant: GlaxoSmithKline (GSK) Biologicals
Date: March 10, 2008
From: Jingyee Kou, HFM-217
Through: A. Dale Horne, Branch Chief, VEB
To:
Laraine Henchal, HFM-478
Luba Vujcic, HFM-475
CC:
Steve Anderson, HFM-210
Henry Hsu, HFM-215
A. Dale Horne, HFM-217
Chron, DCC
Contents
1. Overview
2. Study Rota-023
3. Study Rota-036
4. Study Rota-033
5. Reviewer's Overall Conclusion
1. Overview
In the application for licensure, the applicant GSK has submitted information from several clinical trials. This statistical review covers mainly the results from 3 phase 3 studies:
- Rota-023: a safety and efficacy trial
- Rota-036: an efficacy trial
- Rota-033: a lot consistency trial
2. Study Rota-023:
Title: "A phase 3, double-blind, randomized, placebo-controlled, multi-country and multi-center study to assess the efficacy, safety and immunogenicity of two doses of GSK Biologicals' oral live attenuated human rotavirus (HRV) vaccine in healthy infants"
2.1 Objectives
Primary Safety Objective [rota-023-report-body.pdf]
The primary safety objective was:
- To determine the safety of GSK Biologicals' HRV vaccine with respect to definite intussusception (IS) within 31 days (Day 0 to Day 30) after each HRV vaccine dose in all subjects (N = 60,000).
This objective was reached if:
-
The upper limit of the two-sided 95% Confidence Interval (CI) of the Risk Difference for the percentage of subjects reporting definite IS within 31 days (Day 0 to Day 30) after any dose was below 6/10,000, a limit based on the study sample size and the anticipated IS incidence rate,
and
- There was no statistically significant increase in the percentage of subjects reporting definite IS within 31 days (Day 0 to Day 30) after any dose (the lower limit of the two-sided 95% CI of the Risk Difference had to be below 0).
Primary Efficacy Objective [rota-023-year-1-report-body.pdf]
The primary efficacy objective was:
To determine if two doses of GSK Biologicals' HRV vaccine can prevent severe RV GE (Rotavirus Gastroenteritis) caused by the circulating wild-type RV strains during the period starting from 2 weeks after Dose 2 until one year of age in the efficacy subset (N = 20,000).
Assuming a 1.5% incidence of severe RV GE in the placebo group during the observation period, and a 70% vaccine efficacy, the pre-specified sample size of 20,000 subjects had at least 80% power to detect a lower limit of the 95% CI for the vaccine efficacy above 50%.
Select definitions:
Severe GE: An episode of diarrhea with or without vomiting that required hospitalization and/or re-hydration therapy (equivalent to WHO plan B or C) in a medical facility.
Severe RV GE: An episode of severe gastroenteritis occurring at least two weeks after the full vaccination course in which rotavirus other than vaccine strain was identified in a stool sample collected during the episode of severe gastroenteritis.
2.2 Study design
This study was designed as a randomized, double-blind, placebo-controlled, multi-country and multi-centre study conducted in 12 countries (11 countries in Latin America and Finland). Subjects were randomly assigned (1:1 randomization ratio) to one of the two parallel groups, HRV vaccine group or Placebo control group. A total of 60,000 subjects were planned to be enrolled in this study. All vaccinated subjects were followed for safety at least until Visit 3.
Graphic illustrations of the study design are presented for subjects followed only for safety from Visit 1(Dose 1) until Visit 3 (40,000 subjects planned) and for subjects followed for safety and efficacy from Dose 1 until Visit 3 and beyond (20,000 subjects planned, Subset A). [From rota-023-report-body.pdf submitted by the applicant.]
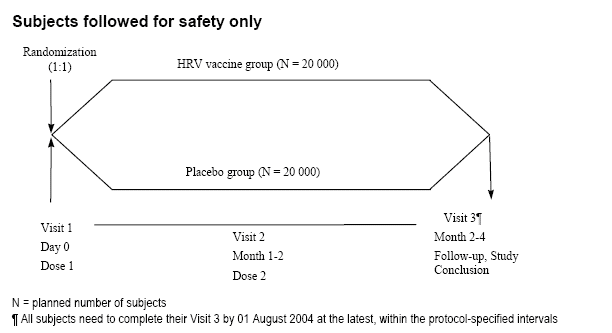
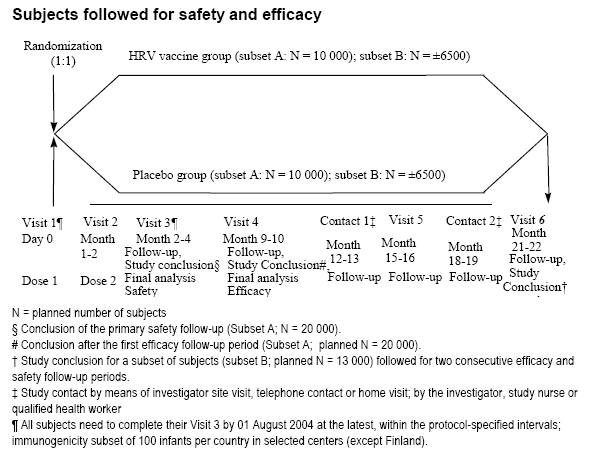
2.3 Sample size for safety evaluation
In the original protocol, the primary safety objective was defined as: "With an assumed background rate of 3 IS cases per 100,000 in the placebo group during the observation period, 60,000 subjects will allow exclusion of an IS attributable risk greater than 2 : 10,000 vaccinees (observed attributable risk ≤ 1 : 10,000 vaccinees, upper limit of 90% CI ≤ 2 : 10,000 vaccinees, at least 80% power)."
While the trial was ongoing, it was determined that the previously assumed background rate was much higher than 3/100,000. A different study estimated the background rate to be about 51/100,000. Consequently, the primary objective for safety was revised to its final form.
The applicant provided the following statements concerning the changes in of the primary objective: [rota-023-report-body.pdf]
"As of 18 May 2004, a total of 14 IS cases were observed within 31 days post vaccination period. This led to an overall IS incidence rate between 2 and 4/10,000, which far exceeded the anticipated definite IS incidence rate of 0.3/10,000 subjects that was expected to occur in the Placebo group in this same time window. This higher incidence of IS could reasonably be attributed to geographical differences and/or the active surveillance for IS in the study.
The higher IS incidence was further substantiated by a concurrent, prospective, multicenter epidemiological study conducted in the same 11 Latin American countries as those participating in study 023. Study epi-204 assessed the incidence of IS through active surveillance in children less than 2 years of age and not vaccinated with HRV. An interim analysis of the epidemiological study showed that most IS cases occurred before one year of age. IS hospitalization was uncommon before two months of age, but increased from three months and peaked at five months of age. Preliminary calculation of background incidence rates in children < than 1 year suggested an overall incidence of 51/100 000, with a range among countries [Study Report 99910/204; Breuer, 2004].
Due to the higher overall IS incidence (study remained blinded) the width of the CI of the Risk Difference had become so large that, under identical IS incidences in both study arms (HRV vaccine minus Placebo), the upper limit of the 90% CI exceeded the initially specified 2/10 000 limit. Therefore the original criterion for meeting the co-primary safety objective was no longer appropriate.
For this reason, the primary safety objective was revised (see Section 5.8.1, amendment 3) so that a vaccine with an identical IS incidence as placebo would meet the objective:
- The upper limit of the two-sided 95% confidence interval of the Risk Difference for definite IS occurring within 31 days post vaccination should be below 6/10 000, a limit based on the study sample size and the anticipated IS incidence rate.
- There should be no statistically significant increase in the incidence of definite IS occurring within 31 days post vaccination (the lower limit of the two-sided 95% CI of the Risk Difference should be below 0)."
The applicant provided the following concerning the definition and analysis of vaccine efficacy:
"The vaccine efficacy was calculated using the formula: 1 - RR = 1 - (ARV/ARU), where RR = relative risk = ARV/ARU
ARU = disease attack rate in unvaccinated population (estimated from the Placebo group) = number of subjects reporting at least one severe RV GE episode / total number of subjects in the placebo (control) group.
ARV = disease attack rate in vaccinated group = nv/Nv = number of subjects reporting at least one severe RV GE episode / total number of subjects in the HRV vaccine group.
Two-sided Fisher's exact test (significance level of a = 0.05) was used to compare these percentages between HRV and Placebo groups."
2.4 Results from Applicant on IS cases
A total of 63,225 infants (31,673 in vaccine group and 31,552 in placebo group) were enrolled and vaccinated in 11 countries in Latin America and Finland for this trial.
Rotarix is a rotavirus vaccine to be administered in two doses. The applicant has provided the following results for definite IS diagnosed within 31 days (Day 0 to Day 30) after any dose. There were 6 cases in the vaccine group and 7 cases in the placebo group within the 31 days after either dose.

[rota-023-report-body.pdf, Table 18]
The applicant concluded that since the upper limit of the 95% CI for the relative difference is < 6/10,000, the revised primary objective for safety has been demonstrated.
2.5 Reviewer's Comments and Analysis on IS cases
- Study Rota-023 was performed outside the US and was not under the US FDA Investigational New Drug (IND) regulation. Therefore, FDA did not have the opportunity to concur with the study plan before or during the study.
- The study was designed with the assumption of a background rate of IS in placebo group of 3/100,000. Because another study obtained an estimate of 51/100,000, together with the number of accumulating IS cases observed during the trial of Rota-023, the primary objective was revised during the conduct of the study. Since changing the primary objective while the trial is ongoing could potentially compromise the integrity of the study, and CBER did not concur with this change during the study, CBER requested more detailed information from the applicant to ensure that proper procedure was followed. The applicant submitted the response in amendment #17 dated February 1, 2008. Because the process was approved by the Independent Data Monitoring Committee (IDMC), it is considered acceptable by the reviewer.
-
The applicant presented the definite IS cases within 31 days (Day 0 to Day 30) by the diagnostic date, not the start date of the symptoms. However, there was one case in the vaccine arm for which the symptoms started on Day 29 but was not diagnosed until Day 31, and, hence, was excluded from the reporting period. The following table, created by the reviewer, displays the IS cases by the onset day of the symptoms for all individuals during the 31-day window.
Last dose
Days since last dose when an IS case occurred
Ratio of the number of cases
Risk Difference (95% CI) Per 10,000
Relative Risk (95% CI) Per 10,000
Rotarix ( N=31673)
Placebo (N=31552)
Dose 1 (day 0 – day 30)
18
16, 22
1 : 2
Dose 2 (day 0 – day 30)
3, 3, 16, 17, 25, 29
6, 9, 18, 24, 28
6 : 5
Any Dose
7 : 7
- 0.008
(-2.63, 2.61)0.996
(0.36, 2.72)From the results obtained by the reviewer, the upper limit of the 95% CI for risk difference is 2.61, which is still below the revised criterion of 6/10,000. Hence, the revised primary objective was achieved.
CBER considers the relative risk as a measure for assessing adverse events for preventive vaccines. The rationale for this preference is that the risk difference, which may be a useful metric for public health policymakers (e.g., determining how many new hospital beds are needed), would tend to minimize the risk of uncommon adverse events associated with vaccination. Since preventive vaccines will potentially be given to many millions of healthy individuals, it is important not to minimize any potential risk.
The reviewer calculated the upper limit of the 95% CI for the relative risk to be 2.72, which may be considered acceptable.
-
The following table, created by the reviewer, displays the onset day of the symptoms that lead to all IS cases after any dose at all times during the follow-up period. There is no apparent pattern for when the IS cases occurred after each dose.
Days since last dose
when an IS case occurredRatio of the number of cases
Rotarix
(N = 31673)Placebo
(N = 31552)Dose 1 (day 0 - day 30)
18
16, 22
1 : 2
Dose 1 (day 31 +)
53
41, 51, 68, 74, 81, 224
1 : 6
Dose 2 (day 0 - day 30)
3, 3, 16, 17, 25, 29
6, 9, 18, 24, 28
6 : 5
Dose 2 (day 31 +)
56, 68, 86, 144, 231
35, 46, 50, 106, 126, 127, 222
5 : 7
13 : 20
-
Since the risk of IS appeared to be increased among recipients of RotaShield during the 3- to 14-day period after the first dose and during the 3- to 7-day period after the second dose, the reviewer created the following table, displaying the days of IS cases for the periods of 3-7 days and 3-14 days.
There were no IS cases post dose 1. Although there were 2 cases in the vaccine arm versus 1 case in the placebo arm for the period of 3-7 days after the second dose, the period of 3-14 revealed 2 cases in each arm. Due to the small number of cases within these periods of time, one cannot rule out that they occurred on these days by chance alone.Days since last dose
when an IS case occurredRatio of number of IS cases
(Rotarix : Placebo)Rotarix
( N = 31673)Placebo
(N = 31552)Dose 2 (Day 3 - Day 7)
3, 3
6
2 : 1
Dose 2 Day 3 - Day 14)
3, 3
6, 9
2 : 2
2.6 Results from Applicant on Efficacy
Vaccine efficacy analysis was performed on the According To Protocol (ATP) cohort which included all subjects from the ATP safety cohort and who received 2 doses of either the investigational vaccine or the placebo, had follow-up beyond 2 weeks after Dose 2 through the end of the first efficacy follow-up period, and had no vaccine strain in stool samples collected between the day of Dose 1 administration and 2 weeks after Dose 2 was administered.
There were 17,867 subjects (9,009 in the investigational vaccine group and 8,858 in the Placebo group) included in the ATP efficacy cohort.
The following table showing efficacy results was submitted by the applicant [rota-023-year-1-report-body.pdf, Table 12].
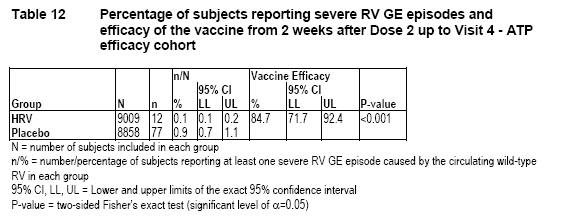
Since the lower bound of the 95% CI for vaccine efficacy is above 50%, the applicant concluded that the primary efficacy objective was reached.
The applicant also used the Cox proportional-hazard model to estimate vaccine efficacy against severe RV GE caused by the circulating wild-type as 84.8% (95% CI: 72.0%; 91.7%). The applicant submitted the following table in amendment # 17 (dated February 1, 2008) at CBER's request.
Table 1 Percentage of subjects reporting severe RV GE episodes and efficacy of the vaccine from 2 weeks after Dose 2 up to Visit 4, by COX - ATP cohort for efficacy
| n/T | Vaccine Efficacy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T | 95%CI | 95%CI | ||||||||
| Group | N | n | (year) | value | LL | UL | % | LL | UL | P-value |
|
Severe RV GE of any wild gtype |
||||||||||
| HRV | 9009 | 12 | 5914.1 | 0.002 | 0.001 | 0.004 | 84.8 | 72.0 | 91.7 | <0.001 |
| Placebo | 8858 | 77 | 5777.1 | 0.013 | 0.011 | 0.017 | ||||
|
G1 wild type |
||||||||||
| HRV | 9009 | 3 | 5916.4 | 0.001 | 0.000 | 0.002 | 91.8 | 73.5 | 97.5 | <0.001 |
| Placebo | 8858 | 36 | 5788.6 | 0.006 | 0.004 | 0.009 | ||||
|
Pooled Non G1 (G2, G3, G4, G9) |
||||||||||
| HRV | 9009 | 10 | 5914.7 | 0.002 | 0.001 | 0.003 | 75.5 | 51.0 | 87.6 | <0.001 |
| Placebo | 8858 | 40 | 5792.3 | 0.007 | 0.005 | 0.009 | ||||
Notes:
N = number of subjects included in each group
n = number of subjects reporting at least one specified severe RV GE episode in each group
T= sum of follow-up period expressed in year censored at the first occurrence of the specified severe RV GE episode, in each group
n/T= person-year rate of the specified severe RV GE in each group
95% CI, LL, UL = Lower and upper limits of the 95% confidence interval
P-value from Cox regression model to test H0 = {VE=0%} (Y = Time to Event)
The numbers of severe RV GE episodes in the ATP cohort by main RV serotypes are displayed in the following table:
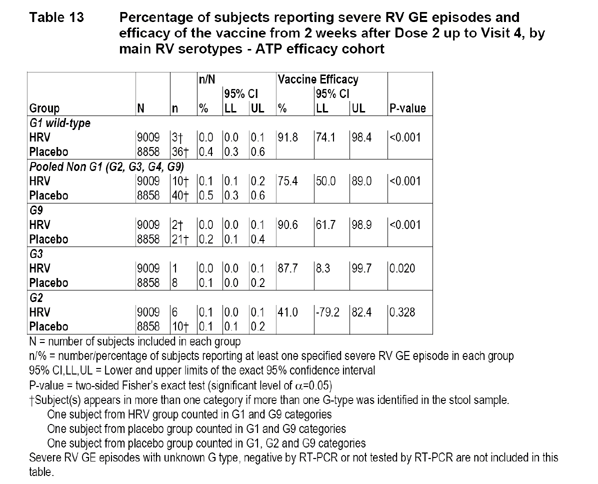
The applicant also provided the efficacy estimate in the Total Vaccinated Cohort which is illustrated in the following applicant-produced table.
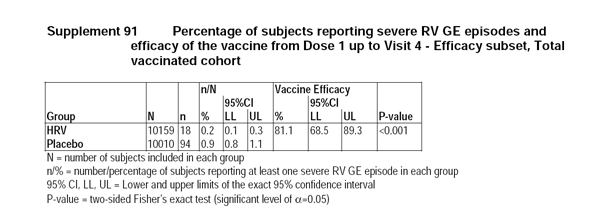
2.7 Reviewer's Comments on Efficacy Analysis for Study Rota-023
- For estimation of vaccine efficacy in this study, CBER prefers to use a "time-to-first-episode" analysis rather than using the number of subjects who had at least one episode among the subjects enrolled in each arm. The rationale for this preference is that in accumulating the denominator of event rates, the time-to-event approach is able to account for differential follow-up times of subjects, while the other approach, which simply accumulates the number of persons enrolled without regard to how long they were under study, does not. Therefore, CBER is inclined to place more importance on the Cox proportional-hazards model results. The result of this analysis was submitted to CBER as an amendment. The reviewer has verified the efficacy estimate and the 95% confidence intervals for any wild-type, as described in the primary objective.
- For individual serotypes in the circulating wild-types, statistically significantly fewer cases were found in G1, G3, and G9 in the HRV group than in the placebo group. However, there were no properly formed hypotheses or power estimates before the trial was conducted. Hence, the observed results are more appropriate for hypothesis forming for future studies than for vaccine label claims.
-
The applicant provided the efficacy estimates on the Total Vaccinated Cohort as 81.1% with a 95% CI (68.5%, 89.3%). Although the reviewer obtained slightly different results from the data submitted by the applicant, the discrepancies do not alter the overall conclusions related to this product.
Group
N
Number of cases (n)
n/N%
Efficacy
(%)95% CI
Lower limit95% CI
Upper limitHRV
10159
19
0.19
80.5
67.9
88.7
Placebo
10010
96
0.96
3. Study Rota-036
Title: "A phase 3b, double-blind, randomized, placebo-controlled, multi-country and multi-center study to assess efficacy, safety and immunogenicity of two doses of GSK Biologicals' oral live attenuatd human rotavirus (HRV) vaccine in healthy infants in co-administration with specific childhood vaccinations"
[From rota-036-report-body.pdf]
3.1 Objectives
Primary objective
The primary objective of this study was:
- To determine the efficacy of two doses of GSK Biologicals' HRV vaccine given concomitantly with specific childhood vaccinations against any RV GE caused by the circulating wild-type RV strains during the first efficacy follow-up period.
3.2 Study design
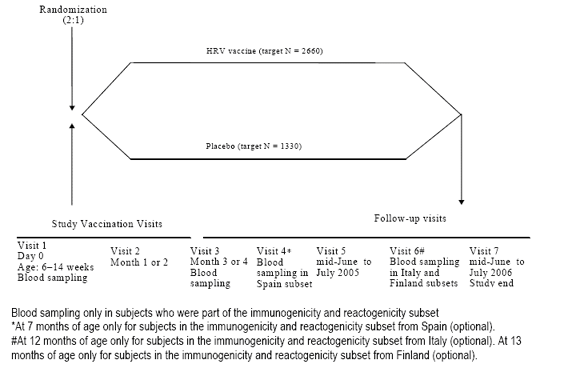
This is a randomized, double-blind, placebo-controlled, multi-country and multi-center study conducted in Czech Republic, Finland, France, Germany, Italy, and Spain. Eligible subjects were randomly assigned (in a 2:1 randomization ratio) to one of the two parallel groups: Group HRV vaccine or Group Placebo (control group).
Subjects in each group were to receive two doses of HRV vaccine or placebo co-administered with the first two doses of the primary childhood vaccination series given according to the national plan of immunization in each country. The third dose of the primary childhood vaccination series was to be administered according to the national plan of immunization in each country.
3.3. Primary Efficacy Endpoint
The primary efficacy endpoint was the occurrence of any RV GE caused by the circulating wild-type RV strains during the first efficacy follow-up period.
An episode of GE will be classified positive for RV caused by the circulating wild-type RV strains if RV other than vaccine strain is identified in a stool sample collected during the episode. A GE episode without stool sample/result available will not be considered in the analysis as a RV GE episode.
RV GE for efficacy analysis is defined as an episode of GE in which rotavirus other than vaccine strain is identified in a stool sample collected during the episode.
The first efficacy follow-up period starts from two weeks after Dose 2 of HRV vaccine or placebo and ends at Visit 5 (when subjects reached approximately one year of age).
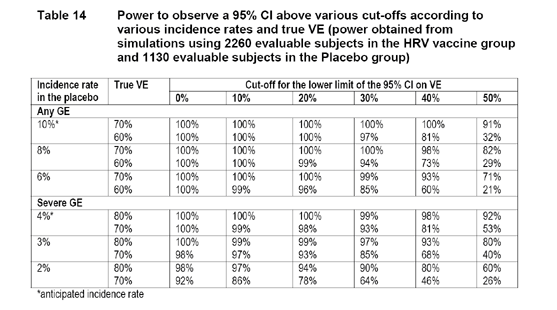
3.4 Sample Size and Power
Considering a 2:1 randomization ratio and various incidence rates, the following table provided by the applicant displays the power for the 95% CI of VE against any RV GE (primary endpoint) to be above given limits. The applicant cited the results from Study Rota-004 in Finland: an incidence rate of 10% for the percentage of placebo recipients with any RV GE caused by the circulating wild-type RV strains during the first efficacy period, which was considered a reasonable assumption. Therefore, if the VE was truly 70%, the study had at least 90% power to observe a 95% CI for the VE that would be above 50%.
Analysis of efficacy
The first efficacy period started from two weeks after Dose 2 of HRV vaccine or placebo and ended at Visit 5. Analysis of efficacy during the first efficacy period was performed on the ATP cohort for efficacy. Analysis of efficacy from the first dose onwards was performed on the total vaccinated cohort.
Only GE episodes in which wild-type RV (i.e., other than the vaccine strain) was identified in a stool specimen were included in the efficacy analysis.
A global overview of the number of GE episodes of any etiology (RV or not) and RV GE episodes reported during the first efficacy period was provided for pooled countries.
Number of GE episodes with no available stool results during the first efficacy period was provided for pooled countries.
For the ATP cohort for efficacy (primary analysis), VE estimates were calculated with their 95% CI against:
- Any RV GE during the period starting from 2 weeks after Dose 2 up to Visit 5.
The VE was defined as the percent reduction in the frequency of the relevant endpoint in vaccinated subjects compared with those subjects who received placebo. This was calculated as follows:
VE = vaccine efficacy = 1- RR = 1 - (ARV/ARU)
where
ARU = disease attack rate in unvaccinated population (estimated from the Placebo group) = nu/Nu = number of subjects reporting at least one RV GE episode / total number of subjects in the Placebo group,
ARV = disease attack rate in vaccinated group = nv/Nv = number of subjects reporting at least one RV GE episode / total number of subjects in the HRV vaccine group,
RR = relative risk = ARV/ARU.
3.5 Applicant's Results
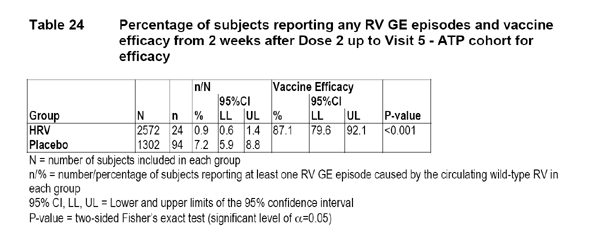
The following table, provided by the applicant, presents the efficacy of the HRV vaccine against any RV GE caused by the circulating wild-type RV during the first efficacy period.
The applicant concluded that significantly fewer subjects in the HRV vaccine group reported any RV GE caused by the circulating wild-type RV compared to the Placebo group (two-sided Fisher's exact P-value < 0.001). VE against any RV GE was 87.1% (95% CI: 79.6%; 92.1%). The primary efficacy objective of the study was reached since the lower limit of the 95% CI for the vaccine efficacy was above 50% (criteria specified for fulfilling the primary efficacy objective).
The applicant also performed an analysis using the Cox proportional-hazard model. The applicant concluded that VE against any RV GE caused by the circulating wild-type RV during the first efficacy period was 87.4% (95% CI: 80.3%; 91.9%). However, details of these results were not submitted with the application.
The numbers of any RV GE episodes in the ATP cohort by main RV serotypes are displayed in the following table:
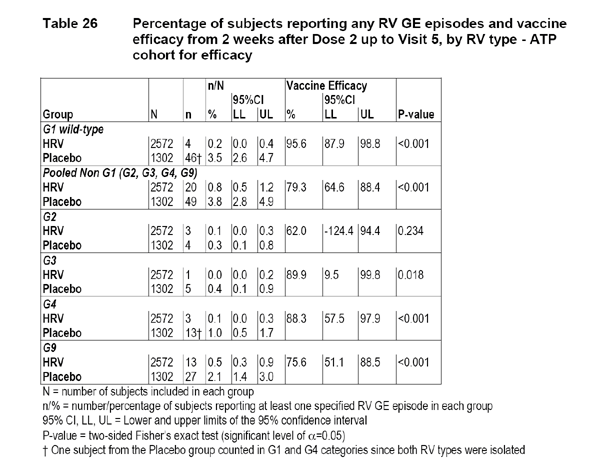
The applicant provided the following table to display the efficacy estimates for the vaccine from dose 1 up to visit 5 in the Total Vaccinated Cohort.
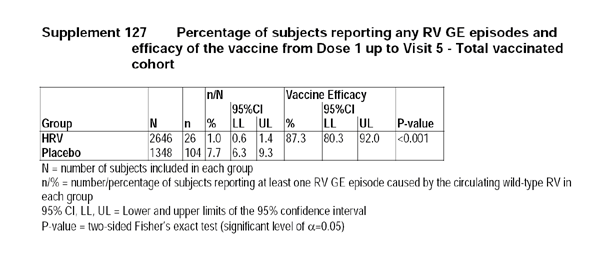
3.6 Reviewer's Comments for Study Rota-036
- This study was not conducted under US FDA IND regulation. Although no specific hypothesis was proposed, the applicant did provide a sample size and power calculation based on the expected incidence rate and proposed efficacy.
- Although no details for the efficacy estimates using the Cox proportional-hazard model were submitted with the application, the results are similar to the ones obtained in a larger phase 3 study, Rota-023. Hence, they are considered acceptable.
- For individual serotypes in the circulating wild-types, statistically significantly fewer cases were found in G1, G3, G4, and G9 in the HRV group than in the placebo group. However, there were no properly formed hypotheses or power estimates before the trial was conducted. Hence, the observed results are more appropriate for hypothesis forming for future studies than for vaccine label claims.
4. Study Rota-033
Title: "A phase 3, randomized, double-blind and placebo-controlled study to assess the clinical consistency of three production lots of GSK Biologicals' HRV vaccine in terms of immunogenicity and safety when given to healthy infants at 2 and 4 months of age"
[From rota-033-report-body.pdf]
4.1 Objectives
Primary objective
The primary objective of this study was:
- To demonstrate the lot-to-lot consistency of the HRV vaccine in terms of immunogenicity as measured by serum anti-rotavirus IgA antibody levels two months after Dose 2.
Consistency would be reached if, for all pairs of lots, the two-sided 90% confidence intervals (CIs) for the ratio of anti-rotavirus IgA antibody Geometric Mean Concentrations (GMCs) two months after Dose 2 are within the [0.5; 2] clinical limit interval.
4.2 Study design
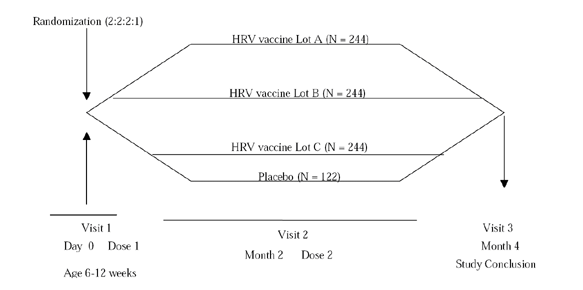
This study had four study groups: three HRV vaccine groups for three consecutive lots of HRV vaccine to test lot-to-lot consistency and a placebo group as the control. Since no comparisons were to be performed between HRV groups and the placebo group to demonstrate a study objective, a lower randomization ratio was used for the placebo group.
4.3 Protocol Amendments/Modifications
There were two amendments to the study protocol. The rationale for each amendment and any major changes to the conduct of the study are described below.
- The protocol was amended on May 23, 2003 before study start. The original protocol planned to test a modified formulation of the HRV vaccine. The amendment reverted to the use of the initial vaccine formulation instead, since non-inferiority of the modified formulation was not established as compared to the initial formulation. The amendment deferred the routine OPV doses from the study vaccine dose by 2 weeks, since OPV can affect immunogenicity of other orally administered vaccines when given simultaneously. To further characterize the rotavirus shedding, the amendment mandated collection of stool samples at specific time points from a subset of subjects. The volume of blood samples to be collected was increased from 1 ml to 2 ml to have sufficient quantity of serum to allow bridging of the two anti-rotavirus IgA assay methods used in studies with GSK Biologicals' HRV vaccine.
- The protocol was amended on January 22, 2004 after all subjects had been enrolled to clarify that the anti-rotavirus IgA antibody concentration in serum samples would be measured by ELISA at Dr. Ward's Laboratory, Children's Hospital Medical Center, Cincinnati, OH, USA. The assay cut-off was 20 units/ml (U/ml).
4.4 Applicant's Results
The applicant provided the following table, depicting the number of subjects enrolled and reasons for exclusion from the According-to-Protocol cohort.
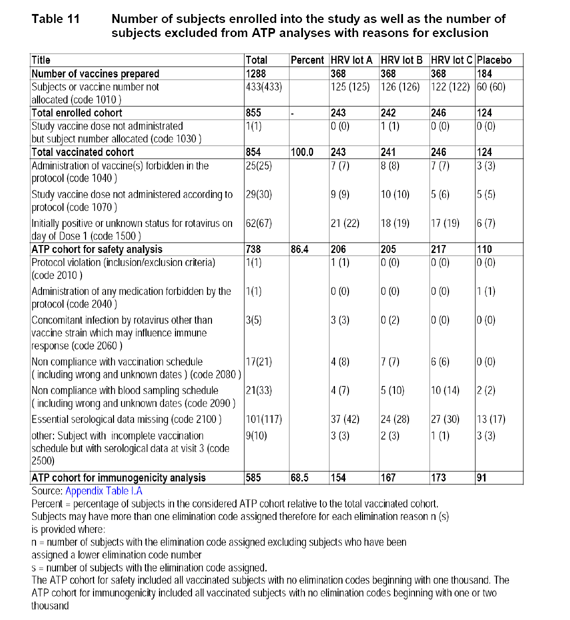
4.5 Applicant's Results
The According-to-Protocol (ATP) cohort for immunogenicity analysis included a total of 585 subjects (154 subjects in Group HRV vaccine lot A, 167 subjects in Group HRV vaccine lot B, 173 subjects in Group HRV vaccine lot C, and 91 subjects in the placebo group).
The following table provided by the applicant displays the GMC and the 95% CI for each of the three HRV lot groups, the pooled 3 lots, and the placebo group.
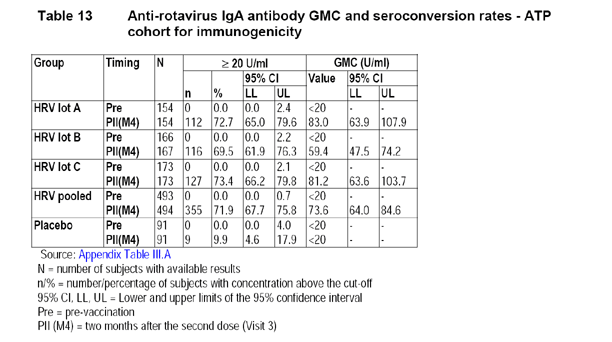
The following table provided by the applicant displays the ratios of GMC of all paired comparisons between 2 of the 3 lots and their 90% CI, respectively.

Because more than 5% of the Total-Vaccinated-Cohort (TVC) was excluded from the According-to-Protocol (ATP) analysis, the applicant provided the following table, which displays the GMC and 95% CI for each of the lots, the pooled, and the placebo group based on the TVC.
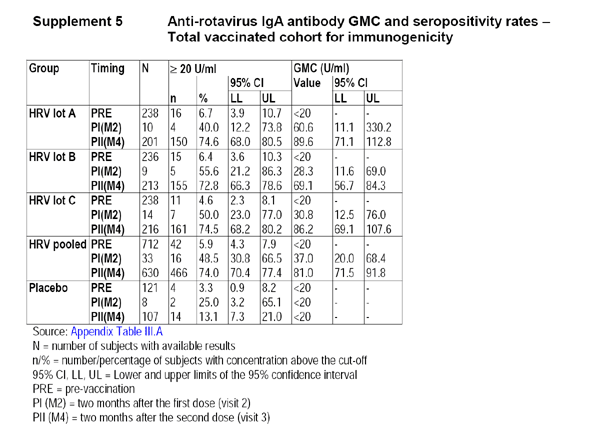
4.6 Applicant's Conclusions
Based on the results found in this study, the applicant suggests:
- Clinical consistency of three consecutive production lots of GSK Biologicals' HRV vaccine was demonstrated in terms of ratios of serum anti-rotavirus IgA antibody GMCs two months after the second dose.
- Immunogenicity results of the total vaccinated cohort were consistent with results for the ATP cohort, indicating that no bias was introduced in the selection of subjects who complied with the per protocol analysis.
4.7 Reviewer's Comments on Study Rota-033
-
This study was not performed under US FDA IND regulation. Therefore, CBER could not provide comments for the applicant before or during the trial. For lot consistency trials, in order to be consistent with use of 95% CI for all other evaluations, CBER prefers the 95% CI to the 90% CI as performed by the applicant. The following table produced by the reviewer displays the ratios and the 95% CI for the paired comparisons of GMCs.
|
Group |
N |
GMC |
Group |
N |
GMC |
Groups |
Ratio |
95% CI |
95% CI |
|---|---|---|---|---|---|---|---|---|---|
|
Lot A |
154 |
83.0 |
Lot B |
167 |
59.4 |
Lot A over Lot B |
1.40 |
0.99 |
1.97 |
|
Lot A |
154 |
83.0 |
Lot C |
173 |
81.2 |
Lot A over Lot C |
1.02 |
0.72 |
1.46 |
|
Lot B |
167 |
59.4 |
Lot C |
173 |
81.2 |
Lot B over Lot C |
0.73 |
0.53 |
1.02 |
Since the upper limits of the 95% CI of the GMCs comparing lots are below 2, the reviewer concurs with the applicant's conclusion that there is lot consistency.
5. Reviewer's Overall Conclusion
- All three phase 3 studies, Study Rota-023, Study Rota-036, and Study Rota-033, were not conducted under US/FDA IND regulation. Therefore, none of the protocols and amendments were concurred upon by CBER before or during the trial.
- By the reviewer's relative risk calculation based on the onset day of the IS symptoms, the applicant has reached the revised primary safety objective.
- The efficacy results based on the Cox proportional-hazard model were confirmed by the reviewer for Study Rota-023. The reviewer considers the Cox model to be a more appropriate method for estimating the efficacy of this vaccine, compared to the applicant's method, due to large variations in the follow-up times of the subjects. The Cox model results from Study Rota-036 are similar to the ones from the larger study, Rota-023, are considered acceptable.
- For individual serotypes in the circulating wild-types, statistically significantly fewer cases were found in G1, G3, G4, and G9 in the HRV group than in the placebo group in separate studies. However, there were no properly formed hypotheses or power estimates before the trial was conducted. Hence, the observed results are more appropriate for hypothesis forming for future studies than for vaccine label claims.
- Lot consistency based on three lots was demonstrated, with upper limits of both the 90% and 95% confidence intervals being under 2.
- From the study results shown above, the reviewer concludes that the applicant has fulfilled the primary objective of each of the studies reviewed here. Therefore, the reviewer concludes that this product may be approved, unless there are other considerations beyond those reviewed here that would warrant otherwise.