

GUIDANCE DOCUMENT
Early Collaboration Meetings Under the FDA Modernization Act (FDAMA); Final Guidance for Industry and for CDRH Staff February 2001
- Docket Number:
- FDA-2020-D-0957
- Issued by:
-
Guidance Issuing OfficeCenter for Devices and Radiological Health, Office of Product Evaluation and Quality
Document issued on: February 28, 2001
This document supersedes: Early Collaboration Meetings Under the FDA Modernization Act (FDAMA); Guidance for Industry and CDRH Staff, February 19, 1998)

U.S. Department of Health and Human Services
Food and Drug Administration
Center for Devices and Radiological Health
Program Operations Staff
Office of Device Evaluation
Center for Devices and Radiological Health
Preface
Public Comment
Comments and suggestions may be submitted at any time for Agency consideration to Dockets Management Branch, Division of Management Systems and Policy, Office of Human Resources and Management Services, Food and Drug Administration, 5630 Fishers Lane, Room 1061, (HFA-305), Rockville, MD, 20852. When submitting comments, please refer to the Docket number 98D-0078 and the exact title and date of this guidance document. Comments may not be acted upon by the Agency until the document is next revised or updated.
For questions regarding the use or interpretation of this guidance contact Thinh Nguyen at 301-796-5560.
Additional Copies
Additional copies are available from the Internet. You may also send an e-mail request to CDRH-Guidance@fda.hhs.gov to receive a copy of the guidance. Please use the document number 310 to identify the guidance you are requesting.
Early Collaboration Meetings Under the FDA Modernization Act (FDAMA);
Final Guidance for Industry and for CDRH Staff
This document is intended to provide guidance. It represents the Agency’s current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind the Food and Drug Administration (FDA) or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statute and regulations.
Guidance on Standard Operating Procedures
FDAMA provides for two early collaboration meetings. These meetings are intended to facilitate interaction between FDA and applicants and provide clear direction for testing and development of those devices requiring clinical investigations to support marketing.
Determination Meeting
A Determination Meeting, as described in §513(a)(3)(D), is available to anyone anticipating submitting a PMA or PDP and is intended to provide the applicant with the Agency’s determination of the type of valid scientific evidence that will be necessary to demonstrate that the device is effective for its intended use. As a result of this meeting, FDA will determine whether clinical studies are needed to establish effectiveness and, in consultation with the applicant, determine the least burdensome way of evaluating device effectiveness that has a reasonable likelihood of success. The applicant can expect that FDA will determine if concurrent randomized controls, concurrent non-randomized controls, historical controls, or other types of evidence will be acceptable. FDA’s determination is to be written, shared with the applicant within 30 days following the meeting, and is binding upon the Agency, unless it would be contrary to public health.
Agreement Meeting
The other opportunity for a meeting established by FDAMA is an Agreement Meeting, described in §520(g)(7), which is open to any person planning to investigate the safety or effectiveness of a class III product or any implant. Thus, unlike the Determination Meeting, the Agreement Meeting is available to submitters of 510(k)s for eligible devices. The purpose of this meeting is to reach agreement on the key parameters of the investigational plan (see 21 CFR 812.25), including the clinical protocol. The meeting is to be held within 30 days of the receipt of a request for such a meeting. Any agreement reached in this meeting is also to be written, shared with the applicant, and made part of the administrative record. It is binding on the Agency and may be changed only with the written agreement of the applicant or when there is a substantial scientific issue essential to determining the safety or effectiveness of the device.
Background
Prior to the passage of FDAMA, the Office of Device Evaluation (ODE) had encouraged IDE sponsors to obtain Agency advice on their proposed bench/animal testing as well as on the design of their clinical trials before submitting an investigational device exemption (IDE) application. Such interactions, referred to as "pre-IDEs," allow the Agency to provide early feedback to sponsors and, in so doing, help to facilitate approval of the IDE. Pre-IDE meetings are informal, and the feedback is non-binding on either party. FDA encourages applicants to continue to take advantage of these informal communications, as outlined in IDE Guidances #D95-1, "Goals and Initiatives for the IDE Program" and #D99-1, "Pre-IDE Program: Issues and Answers." FDAMA, for the first time, provides for formal collaboration meetings (Determination and Agreement) where the results of the meetings are now binding.
The Determination and Agreement Meetings should take place early in the development of the device so the applicant may use the meeting results to plan efficiently for the appropriate study. FDAMA is silent as to whether these early collaboration meetings are individual events or part of an ongoing dialogue with the applicant. Experience and practicality suggest that, particularly with novel device/technologies, prior dialogue is critical to the effectiveness of these formal binding meetings. Preliminary discussions can help clarify and focus on the key issues to be addressed in the pre-submission materials and in the meetings themselves. Preliminary discussions may also help an applicant decide between the use of formal binding meetings and informal meetings. Depending on FDA’s familiarity with the device and the degree to which relevant precedents exist, one or more informal meetings may be useful before a Determination or Agreement meeting can be productive.
A graphical illustration of how early collaboration meetings fit into this communication continuum is presented below. The x-axis represents the device development timeline, while the y-axis represents the amount known about the device.
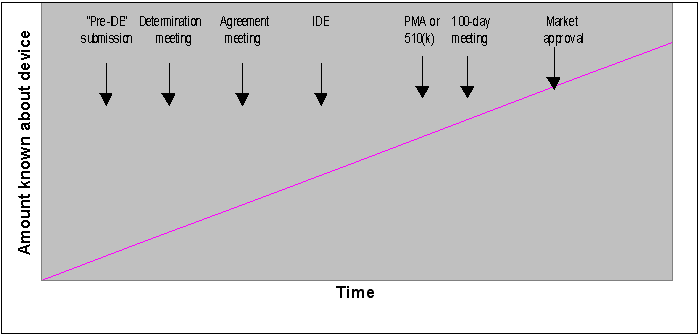
Figure 1: Graphical illustration of a typical approach to communication about a new device. Vertical arrows illustrate typical timing of potential communications between the sponsor and the Agency; not all will be applicable to every device. Informal communications are also encouraged throughout the device development process.
In recognition that the Agency will not be able to reach useful conclusions unless a fair amount of planning and commitment have been undertaken by the applicant prior to the meeting, FDAMA requires that the applicant submit a formal package to the Agency at the time of the request. According to FDAMA, the applicant requesting a meeting shall include: (1) a detailed description of the device; (2) a detailed description of the proposed conditions of use; (3) a proposed plan for determining whether there is a reasonable assurance of effectiveness; (4) if available, information regarding the expected performance of the device; and (5) a detailed clinical protocol (for the Agreement Meeting).
With preliminary discussions and good planning, a Determination or Agreement Meeting should be completed in one session. While it is expected that well-prepared attendees will normally reach consensus on the substantive issues during the course of the meeting, there may be occasions when new issues emerge from the discussion. For this reason, it is important that the applicant firm be represented by someone who has the authority to speak for the firm so that agreement on these new issues may be reached. Infrequently, unresolved issues may remain at the end of the meeting. In these exceptional situations, the applicant may request that the issue(s) be tabled and the meeting suspended. Upon mutual agreement, the early collaboration meeting would be reconvened at a future time acceptable to both parties to permit additional, relevant information to be considered by FDA staff before a binding determination or agreement is reached.
FDAMA makes it clear that the determinations or agreements resulting from these meetings are to be binding. In the case of a Determination Meeting under §513(a)(3)(D), the determination regarding valid scientific evidence is binding on the Agency and cannot be changed unless FDA concludes that adhering to it could be contrary to public health. Further, the Agency is to specify if clinical data are necessary to establish device effectiveness. In deciding what type of clinical studies should be conducted, if any, the Agency is charged with considering, in consultation with the applicant, the least burdensome way of evaluating device effectiveness that has a reasonable likelihood of success. In the case of an Agreement Meeting under §520(g)(7), the agreement is binding on the Agency. FDAMA specified that the Agency may only change the agreement when a substantial scientific issue essential to determining the safety or effectiveness of the device has been identified, and only following an opportunity for the applicant to meet with FDA to discuss the scientific issue involved.
The binding nature of the agreement or determination is predicated on the applicant not significantly changing the bases of the agreement or determination (e.g., intended use and indications, product design, investigational plan, clinical study protocol, etc.). If these bases are significantly changed, then the agreement or determination will have been abrogated and the Agency's agreement or determination will no longer be in effect.
Finally, it should be noted that the two new statutory provisions do not address other pre-submission meetings or topics outside the scope of the agreement or determination. That is, the meetings are not intended to serve as pre-PMA submission meetings to discuss formatting of the application or the strength of a particular clinical data set already produced by a potential PMA applicant. Pre-PMA submission meetings to address these types of issues should continue to be held; however, FDA staff should clarify to the applicant that the meeting is intended to help guide the PMA submission, but it is not a FDAMA-mandated meeting that will result in a binding agency commitment. Similarly, the Agreement/Determination Meetings should stay focused on the relevant issues at hand. Other topics, such as the value of a feasibility study or the type of data needed to support a labeling claim, may arise. Because such discussions are outside the scope of this type of meeting, these discussions should be left to the informal meetings.
Determination Meeting
Applicant Activities
Meeting Preparation
- Contact the appropriate branch to have a general discussion regarding the device.
- A request for a Determination Meeting should be: a) clearly identified as such and b) submitted in triplicate to the Document Mail Center (HFZ-401). The Agency expects that the submission will be approximately 10-20 pages in length and include the following items:
- Description of the device, highlighting elements involving new technologies or raising new clinical issues. This description should include, at a minimum:
- General device description
- Engineering drawing
- Device composition/key components/materials
- Mechanism of action/principles of operation
- Device specifications
- Analysis of potential failure modes
- Outline of the proposed conditions of use, including:
- Proposed intended use and indications
- Population for whom use is indicated and environment of use, if applicable
- Summary of instructions for use of the product
- Any proposed warnings, precautions, contraindications, restrictions or training requirements, if known
- Proposed plan for clinical evaluation of the product (if needed), including:
- Summary of the risk analysis
- Primary endpoints/objectives and how they will be measured
- Secondary endpoints/objectives and how they will be measured
- Success/failure criteria for the overall study and both primary and secondary individual endpoints
- Study design - controlled/uncontrolled, prospective/retrospective, randomized/nonrandomized and justification for study design
- Type of control (historical, concurrent, active, none, etc.), identification of control, and rationale or justification of the control
- Relevant performance information on the device, especially if the device involves new technology or raises new clinical issues. This information may include:
- Published and/or unpublished data
- Summary of bench and/or animal test data
- Summary of prior clinical experience
- Description of the device, highlighting elements involving new technologies or raising new clinical issues. This description should include, at a minimum:
- Arrange for a preliminary meeting (videoconference or teleconference) to review the submitted information, identify key issues, and discuss any additional information that may need to be submitted prior to the actual Determination Meeting.
- Submit additional information by hardcopy and email or facsimile, as required, and arrange date and time for formal Determination Meeting.
The Meeting
Meetings may be face-to-face, by videoconference, or by teleconference and should be scheduled for approximately 1-2 hours. It is important to note that the meeting format will generally be different for a pre-meeting than for the formal meeting. At a pre-meeting, the applicant should present and discuss the material submitted to the Agency. The Agency attendees should raise any questions for the applicant and request any additional information to be submitted prior to the formal meeting. Finally, the key issues to be addressed in the determination should be identified. At the formal meeting, the discussion should be focused on these key issues, leading to the Agency’s determination.
Post-Meeting Activities
- The applicant should draft meeting minutes and exchange these with the Agency for review/comment within 7 days following the meeting.
- The applicant may wish to complete the checklist entitled, "The Sponsor’s Evaluation of the Application of the Least Burdensome Principles in Early Collaboration Meetings." The Agency will use this checklist to help assess the industry’s satisfaction with FDA’s application of the least burdensome approach to determining the type of valid scientific evidence needed for marketing approval. The form may be found in Appendix B and, once completed, it may be faxed to Ms.Wanda Sawyer-Major, Program Operations Staff at (301) 594-2977 at anytime.
Agency Activities
Meeting Preparation
- A request for a Determination Meeting will be logged in as a pre-IDE submission for tracking purposes (flagged as a Determination Meeting Request) and assigned a number (e.g., I000001). Within 30 days of receipt, the team leader for the submission will contact the applicant to establish dates for any pre-meetings and the formal Determination Meeting.
- The review division will decide which FDA staff should attend the meeting, but may include persons specifically requested by the applicant. The attendees may include: the team leader/project manager, medical officer, statistician, other scientists with expertise in the product area (from ODE, Office of Compliance (OC), Office of Surveillance and Biometrics (OSB), and/or Office of Science and Technology (OST)), the appropriate branch chief, a division associate, deputy or director, and a member of the Program Operations Staff (POS). For meetings likely to consider novel products, development strategies, or controversial issues, the division should discuss the issue with and, if appropriate, include the participation of the Director or Deputy Director of ODE.
- FDA should conduct an internal pre-meeting (more than one may be necessary) to ensure that everyone is familiar with the issues. In accordance with "The Systems Approach," this meeting should also be used to consider our knowledge of and experience with similar products and to formulate FDA’s overall strategy for addressing the situation. A member of POS should be invited to the internal pre-meeting to provide background information on similar issues faced by other divisions and to offer guidance on any unique or controversial regulatory issues raised by the applicant’s request. The attendees should identify questions and issues to be discussed with the applicant. The timeline for any pre-meetings and the formal meeting with the applicant should also be discussed.
The Meeting
See the section with this title on the previous page.
Post-Meeting Activities
- At the end of the meeting, the Agency team leader for the application will summarize the determination or explain any arrangements for tabling the determination, including the date of the next meeting, if appropriate. A record of attendees and minutes of the meeting should be kept by both a designated FDA and applicant attendee. A laptop computer may be helpful in recording the issues discussed and the determination reached. FDA and the applicant should exchange their respective meeting minutes for review following the meeting. This exchange may occur electronically for efficiency. The minutes should be in sufficient detail to reflect the substance of the issues discussed at the meeting; a bulleted format may be helpful.
- The Agency team leader should prepare the memorandum of Agency determination. POS will work with the team leader and review division to develop the memorandum, and in so doing, help to ensure that it accurately reflects the outcome of the meeting and is consistent with Office policy. Within two weeks of the meeting, a draft of the memorandum should be circulated for review among the FDA participants. The memorandum will then be signed by the Division Director and conveyed to the applicant within 30 days of the meeting. In addition, the Agency team leader should complete the checklist entitled, "FDA’s Evaluation of the Application of the Least Burdensome Principles in Early Collaboration Meetings." The checklist can be found in Appendix A of this document and is intended to be used to evaluate whether the least burdensome principles were considered in reaching the Agency determination. A copy of the memorandum and checklist should be placed in the document jacket, and the pre-IDE should be logged out by the division and POS.
1 "A Systems Approach to Premarket Review" was issued on July 17, 2000 and may be found on CDRH’s website.
Agreement Meeting
Applicant Activities
Meeting Preparation
- Contact the appropriate branch to have a general discussion regarding the device.
- A request for an Agreement Meeting should be: a) clearly identified as such and b) submitted in triplicate to the Document Mail Center (HFZ-401). The submission should include the information described above for Determination Meetings as well as the clinical protocol. Any particular items upon which agreement is to be reached should be clearly identified. These items may include:
- Sample Size, including a statistical justification
- Inclusion/Exclusion Criteria
- Endpoints (Primary and Secondary with acceptance/failure criteria)
- Overall study success criteria including justification
- Study duration
- Number of study sites
- Data Monitoring Committee (DMC) operations (these are also sometimes called Data and Safety Monitoring Boards (DSMBs))
- Planned statistical analysis
- Special informed consent provisions, e.g., waivers
The Agency expects that the submission should be approximately 10-20 pages in length.
- Arrange for a preliminary meeting (videoconference or teleconference) to review the submitted information and to identify key issues that the applicant wishes to reach agreement on with the Agency. It is critical that the applicant be as specific as possible (e.g., sample size) in identifying the issues for agreement. Identifying broad concepts (e.g., the clinical protocol) may make reaching an agreement more difficult given the number of variables that would need to be addressed. Finally, the applicant should determine if any additional information needs to be submitted prior to the actual Agreement Meeting.
-
Submit additional information, as required, and provide a list of acceptable dates and times for the formal Agreement Meeting.
The Meeting
Meetings may be face-to-face, by videoconference, or by teleconference and should be scheduled for approximately 1-2 hours. It is important to note that the meeting format will generally be different for a pre-meeting than for the formal meeting. At a pre-meeting, the applicant should present and discuss the material submitted to the Agency. The Agency attendees should raise any questions for the applicant and request any additional information to be submitted prior to the formal meeting. Finally, the key issues on which agreement is to be reached should be identified by the applicant. At the formal meeting, the discussion should be focused on these key issues, hopefully, resulting in a number of areas of agreement.
Post-Meeting
- The applicant should draft meeting minutes and exchange these with the Agency for review/comment within 7 days following the meeting.
- The applicant may wish to complete the checklist entitled, "The Sponsor’s Evaluation of the Application of the Least Burdensome Principles in Early Collaboration Meetings." This checklist will be used by the Agency to help assess the industry’s satisfaction with FDA’s application of the least burdensome approach in its discussion of clinical trial design issues during the Agreement Meeting. The form may be found in Appendix B, and, once completed, it may be faxed to Ms.Wanda Sawyer-Major, Program Operations Staff at (301) 594-2977 at anytime.
Agency Activities
Meeting Preparation
- A request for an Agreement Meeting will be logged in as a pre-IDE for tracking purposes (flagged as an agreement meeting request) and assigned a number (e.g., I000001). Within 7 days of receipt, the team leader for the submission should contact the applicant to establish dates for pre-meeting(s) and the formal Agreement Meeting. As specified in §520(g)(7)(A), the formal Agreement Meeting should occur within 30 days of receipt of the request. It is strongly recommended, however, that a mutually agreed-upon timeline be established to increase the probability of a successful meeting. Therefore, the applicant may request deferral of the formal meeting until one or more informal pre-meetings is completed.
- The review division will decide which FDA staff should attend the meeting, but may include persons specifically requested by the applicant. The attendees may include: the team leader/project manager, medical officer, statistician, other scientists with expertise in the product area (from ODE, OC, OSB and/or OST), the appropriate branch chief, a division associate, deputy or director and a member of the POS. For meetings likely to consider novel products, development strategies, or controversial issues, the division should discuss the issue with, and if appropriate, include the participation of the Director or Deputy Director of ODE.
- FDA should conduct an internal pre-meeting (more than one may be necessary) to ensure that everyone is familiar with the issues. In accordance with "The Systems Approach,"2 this meeting should also be used to consider our knowledge of and experience with similar products and to formulate FDA’s overall strategy for addressing the situation. A member of POS should be invited to the internal pre-meeting to provide background information on similar issues faced by other divisions and to offer guidance on any unique or controversial regulatory issues raised by the applicant’s request. The attendees should identify questions and issues to be discussed with the applicant. The timeline for any pre-meetings and the formal meeting with the applicant should also be discussed.
The Meeting
See the section with this title on the previous page.
Post-Meeting
- At the end of the meeting, the Agency team leader for the application will summarize the agreements or explain any arrangements for tabling certain issues, including the date of the next meeting, if appropriate. A record of attendees and minutes of the meeting should be kept by both a designated FDA and applicant attendee. A laptop computer may be helpful in recording the issues discussed and any agreements that have been reached. The FDA applicant should exchange their respective meeting minutes for review following the meeting. This exchange may occur electronically for efficiency. The minutes should be in sufficient detail to reflect the substance of the issues discussed at the meeting; a bulleted format may be helpful.
- The team leader should prepare the memorandum of agreement. POS will work with the team leader and review division to develop the memorandum and, in so doing, help to ensure that it accurately reflects the outcome of the meeting and is consistent with Office policy. Within two weeks of the meeting, a draft of the memorandum should be circulated for review among the FDA participants. The memorandum will then be signed by the Division Director and conveyed to the applicant within 30 days of the meeting. In addition, the Agency team leader should complete the checklist entitled, "FDA’s Evaluation of the Application of the Least Burdensome Principles in Early Collaboration Meetings." This checklist can be found in Appendix A of this document and is intended to be used to evaluate whether the least burdensome principles were considered in reaching the agreement. A copy of the memorandum and checklist should be placed in the document jacket, and the pre-IDE should be logged out by the division and POS.
Appendix A
FDA’s Evaluation of the Application of the Least Burdensome Principles
in Early Collaboration Meetings
Type of Meeting:
___ Determination Meeting §513(a)(3)(D)
___ Agreement Meeting §520(g)(7)
Division/Branch: ______________________________________________________
Application Number: ___________________________________________________
Sponsor: ____________________________________________________________
Device: _____________________________________________________________
In the early collaboration meeting with the sponsor, were the Least Burdensome principles applied in:
- Determining the Need for Prospective Clinical Data?
-
Was pre-clinical testing considered in lieu of clinical data?
___ Yes ___ No
Explain: _______________________________________________________________ _____________________________________________________________________
-
Was the use of previously collected non-U.S. data, literature, and/or registry data considered?
___ Yes ___ No
Explain:_______________________________________________________________ _____________________________________________________________________
-
- Designing the Clinical Trial?
-
Were alternatives to an actively controlled trial considered?
___ Yes ___ No
If yes, check the following:
i. Literature control
___Yes ___ Noii. Historical control
___ Yes ___ Noiii. Non-active control
___ Yes ___ Noiv. Patient as their own control
___ Yes ___ Nov. Objective Performance Criteria
___ Yes ___ Novi. Other
___ Yes ___ NoIf other, describe: ___________________________________________________________
If alternatives could not be used, explain:__________________________________________
_________________________________________________________________________ -
Was the use of surrogate endpoints considered?
___ Yes ___ NoExplain: ________________________________________________________________
________________________________________________________________________ -
Was a least burdensome approach considered in determining how the primary and secondary endpoints will be measured?
___ Yes ___ NoExplain: ________________________________________________________________
_______________________________________________________________________ -
Was early submission of the application considered? That is, could the application be submitted after a mutually agreed to percentage of the patients had been followed for a pre-defined period of time?
___ Yes ___ NoExplain:_________________________________________________________________
________________________________________________________________________ -
Was the role of postmarketing information considered as a mechanism of reducing the premarket requirements?
___ Yes ___ NoExplain:_________________________________________________________________
________________________________________________________________________
-
-
Were the least burdensome principles applied in other areas of the trial design not mentioned above?
___ Yes ___ NoIf yes, describe: _____________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
___________________________________________________________________________
_______________________________________________________________________________________________________________________
Team Leader signature Date
Appendix B
Sponsor’s Evaluation of the Application of the Least Burdensome Principles in Early Collaboration Meetings
Type of Meeting:
___ Determination Meeting §513(a)(3)(D)
___ Agreement Meeting §520(g)(7)
Division/Branch:________________________________________________________
Application Number:_____________________________________________________
Sponsor:_______________________________________________________________
Device:_________________________________________________________________
In the early collaboration meeting with the sponsor, were the Least Burdensome principles applied in:
- Determining the Need for Prospective Clinical Data?
-
Was pre-clinical testing considered in lieu of clinical data?
___ Yes ___ NoExplain: ________________________________________________________________ __________________________________________________________________________
-
Was the use of previously collected non-U.S. data, literature, and/or registry data considered?
___ Yes ___ NoExplain: ________________________________________________________________ ________________________________________________________________
-
- Designing the Clinical Trial?
-
Were alternatives to an actively controlled trial considered?
___ Yes ___ NoIf yes, check the following:
i. Literature control
___ Yes ___ Noii. Historical control
___ Yes ___ Noiii. Non-active control
___ Yes ___ Noiv. Patient as their own control
___ Yes ___ Nov. Objective Performance Criteria
___ Yes ___ Novi. Other
___ Yes ___ NoIf other, describe: ________________________________________________________
If alternatives could not be used, explain:______________________________________ ________________________________________________________________________
-
Was the use of surrogate endpoints considered?
___ Yes ___ NoExplain: ________________________________________________________________ _______________________________________________________________________
-
Was a least burdensome approach considered in determining how the primary and secondary endpoints will be measured?
___ Yes ___ NoExplain: ________________________________________________________________ _______________________________________________________________________
-
Was early submission of the application considered? That is, could the application be submitted after a mutually agreed to percentage of the patients had been followed for a pre-defined period of time?
___ Yes ___ NoExplain:_________________________________________________________________ ________________________________________________________________________
-
Was the role of postmarketing information considered as a mechanism of reducing the premarket requirements?
___ Yes ___ NoExplain:___________________________________________________________________
-
-
Were the least burdensome principles applied in other areas of the trial design not mentioned above?
___ Yes ___ NoIf yes, describe: _____________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________ ___________________________________________________________________________
Applicant Contact Name and Phone Number (Optional):_______________________________
Once completed, fax to Ms. Wanda Sawyer-Major at (301) 594-2977
- Determining the Need for Prospective Clinical Data?
Submit Comments
You can submit online or written comments on any guidance at any time (see 21 CFR 10.115(g)(5))
If unable to submit comments online, please mail written comments to:
Dockets Management
Food and Drug Administration
5630 Fishers Lane, Rm 1061
Rockville, MD 20852
All written comments should be identified with this document's docket number: FDA-2020-D-0957.