

Sterilization Process Controls

Sterilization Process Controls
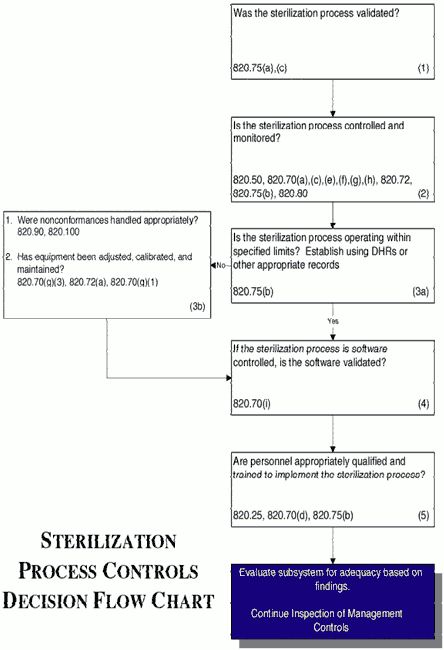
Inspectional Objectives
- Confirm that the sterilization process was validated by reviewing the validation study.
- Review the specific procedure(s) for the sterilization process selected and the methods for controlling and monitoring the process. Verify that the process is controlled and monitored.
- If review of the Device History Records (including process control and monitoring records, acceptance activity records, etc.) reveals that the sterilization process is outside the firm's tolerance for operating or performance parameters:
- Determine whether the nonconformances were handled appropriately; and
- Review the equipment adjustment, calibration and maintenance
- If the sterilization process is software controlled, confirm that the software was validated.
- Verify that personnel have been appropriately qualified and trained to implement the sterilization process.
Sterilization Process Controls
Narrative
Purpose/Importance
The purpose of the production and process control subsystem (including sterilization process controls) is to manufacture products that meet specifications. Developing processes that are adequate to produce devices that meet specifications, validating (or fully verifying the results of) those processes, and monitoring and controlling the processes are all steps that help assure the result will be devices that meet specifications. For sterilization processes, the primary device specification is the desired Sterility Assurance Level (SAL). Other specifications may include sterilant residues and endotoxin levels.
If you are inspecting a contract sterilizer, Inspectional Objectives 2 through 5, described below, are applicable and must be performed. Inspectional Objective 1 regarding validation is applicable only in so far as the contract sterilizer has assumed any responsibility for validation of the process, as indicated in the written agreement between the device manufacturer and the contract sterilizer.
 1. Confirm that the sterilization process was validated by reviewing the validation study.
1. Confirm that the sterilization process was validated by reviewing the validation study.
Validation studies (according to established procedures) are required for sterilization processes.
The review of the sterilization process validation study may be limited to a review of the Validation Study Summary (if available) and Approval if the complete validation study was assessed during the previous inspection and there have been no significant changes in the process, product or package that may impact sterilization effectiveness.
When conducting a complete sterilization process validation study assessment, the items included in the narrative note under Objective 4 of the Production and Process Controls chapter of this Handbook apply. A complete sterilization process validation study assessment must include a review of the established validation procedures and verification (via a review of objective evidence) that: 1. Based upon the bioburden of the product, the defined sterilization process parameters will consistently be effective in obtaining a predetermined Sterility Assurance Level (SAL); and 2. The defined process parameters will not adversely affect product and package performance.
Objective evidence that the sterilization process parameters will consistently be effective in obtaining a predetermined Sterility Assurance Level (SAL) includes records documenting: 1. The determination of product bioburden; 2. The establishment of process parameters and tolerances; 3. The definition of acceptance criteria for a successful validation study; 4. The process challenge studies (e.g. half cycle runs for Ethylene Oxide, verification dose experiments for radiation, or media fills for aseptic processing); and 5. The results of process control and monitoring and acceptance activities (control charts, Biological Indicators, Dosimeters, etc.) used to demonstrate that predetermined acceptance criteria had been met.
 NOTE: Many firms sterilize their products according to the guidance provided within consensus standards (e.g. AAMI/ANSI/ISO standards). These standards are specific to various types of sterilization processes. FDA recognizes many of these standards. This means FDA finds them acceptable. A list of recognized sterilization standards appears at FDA's Center for Devices and Radiological Health (CDRH's) web site.
NOTE: Many firms sterilize their products according to the guidance provided within consensus standards (e.g. AAMI/ANSI/ISO standards). These standards are specific to various types of sterilization processes. FDA recognizes many of these standards. This means FDA finds them acceptable. A list of recognized sterilization standards appears at FDA's Center for Devices and Radiological Health (CDRH's) web site.
Firms may elect to comply with these standards. However, compliance to the standards is voluntary. When a firm claims to comply with one of the recognized standards, the requirements of the standard must be met. If a firm does not claim to comply with a recognized standard, it must provide a scientific rationale supporting the method used for validating and processing its sterilization loads.
Objective evidence that process parameters will not adversely affect product and package performance include records documenting performance testing of the product and packaging following the sterilization process or multiple sterilization processes (if applicable).
Determine whether periodic assessments (e.g. revalidations, sterility dose audits, etc.) of the adequacy of the sterilization process are conducted. Review the records of one periodic assessment of the adequacy of the sterilization process.
NOTE: Many device manufacturers use contract sterilizers for sterilization of their devices. These manufacturers retain the responsibility for the sterility of the finished devices even though sterilization processing is not performed at their own facilities. Therefore, your inspection of a manufacturer that uses the services of a contract sterilizer must verify that the manufacturer has assumed that responsibility. Inspectional Objectives 1 through 3 are applicable in this situation because the manufacturer must be able to provide to you the documentation regarding sterilization validation and processing of its devices regardless of the location of these activities. Although the manufacturer may not have detailed records regarding Objectives 4 and 5 for the contractor's software and personnel, he must have assured the adequacy of these activities by the contractor, through activities such as an audit of the contractor, visits to the contractor, or review of documentation from the contractor. Objective 5 regarding qualifications of the manufacturer's own Q.C. personnel should be covered during your inspection of the manufacturer. 2. Review the specific procedure(s) for the sterilization process selected and the methods for controlling and monitoring the process. Verify that the process is controlled and monitored.
The sterilization process must be validated. However, this does not mean that verification activities utilized to monitor and control the process are unnecessary.
If performed at this location, confirm that the sterilization process, associated environmental and contamination controls, and monitoring and acceptance procedures maintained by the shop floor are the most current approved revision contained within the Device Master Record (DMR). Most firms maintain a "Master List" of the currently approved documents. This list can be verified against the DMR and brought to the shop floor to compare with the currently available documents.
Verify that the building is of suitable design and contains sufficient space to perform necessary operations.
Verify that the control and monitoring activities demonstrate that the process is currently operating in accordance with the DMR. Sterilization parameters which may need to be monitored and controlled include: time, temperature, pressure, load configuration, and humidity. Several of these parameters may require monitoring and control prior to, during and after sterilization processing (e.g. preconditioning, conditioning and aeration in Ethylene Oxide processing). Verification activities used to monitor and control the sterilization process may include: bioburden testing, Biological Indicator (BI) testing, Chemical Indicator (CI) testing, process control record review, sterilant residue testing, and endotoxin testing.
Additionally, packaging integrity verification activities must be reviewed for every inspection during which sterilization is covered. This review of the control and monitoring activities should be done on the shop floor by reviewing work instructions, product acceptance procedures, control charts, etc.
While on the shop floor, make note of one piece of significant sterilization process equipment and one significant piece of inspection, measuring or test equipment (preferably from a finished device acceptance activity). Prior to concluding the inspection, confirm that the applicable maintenance activities (preventive maintenance, cleaning and adjustment, etc.) are performed as scheduled for the chosen piece of sterilization process equipment. Also, confirm that the piece of inspection, measuring, and test equipment was controlled and calibrated.
After you have reviewed the process control and monitoring activities on the shop floor, use the sampling tables and select for review a number of Device History Records (DHRs, including monitoring and control records, acceptance testing records, etc.) from recent production runs. If the process is run over more than one shift, your review should include DHRs from all shifts. Verify that the product was sterilized in accordance with the DMR. Your review of the selected records should include all applicable verification activities (see above) including records of process parameter monitoring, and in-process and final device acceptance activities and results.
Your evaluation must also include a review of the firm's purchasing controls and receiving acceptance activities regarding at least one component, material or service. Examples include: the sterilant, sterilization indicators, and services provided by contract sterilizers or contract laboratories. In addition, review environmental and contamination control records (e.g. bioburden sampling, testing and results). Verify that the sampling plans for process and environmental control and monitoring activities are based upon a valid statistical rationale.
If your review of the Device History Records reveals no anomalies, proceed to Objective 4.
If evidence that the process or environment are not controlled and monitored (no control and monitoring activities, not operating within most currently approved parameters, etc.) is observed, this may be a major production and process control deficiency.
 Important linkages to consider at this point include: Documents, Records and Change Controls (820.180 Records, 820.181 Device Master Record, 820.184 Device History Record, 820.40 Document Controls); Facilities and Equipment Controls (820.72 Inspection, Measuring, and test Equipment); Material Controls (820.50 Purchasing Controls, 820.80 Receiving, In-process, and finished device acceptance, 820.140 Handling, 820.150 Storage, and 820.160 Distribution, and 820.250 Statistical Techniques, 820.60 Identification, 820.65 Traceability, 820.86 Acceptance Status); 820.130 Packaging; and 820.250 Statistical Techniques.
Important linkages to consider at this point include: Documents, Records and Change Controls (820.180 Records, 820.181 Device Master Record, 820.184 Device History Record, 820.40 Document Controls); Facilities and Equipment Controls (820.72 Inspection, Measuring, and test Equipment); Material Controls (820.50 Purchasing Controls, 820.80 Receiving, In-process, and finished device acceptance, 820.140 Handling, 820.150 Storage, and 820.160 Distribution, and 820.250 Statistical Techniques, 820.60 Identification, 820.65 Traceability, 820.86 Acceptance Status); 820.130 Packaging; and 820.250 Statistical Techniques.
3. If review of the Device History Records (including process control and monitoring records, acceptance activity records, etc.) reveals that the sterilization process is outside the firm's tolerance for operating or performance parameters:
- Determine whether the nonconformances were handled appropriately; and
- Review the equipment adjustment, calibration and maintenance
If process or product nonconformance(s) are identified based upon these activities, determine whether the nonconformance(s) were recognized by the firm, handled appropriately and fed into its CAPA system.
Review (if appropriate) the firm's nonconforming product control, review and disposition activities and any CAPA's indicated. If the CAPA included a retest, review the firm's rationale for invalidating the original test results. If the CAPA included resterilization, confirm that the effects of the resterilization process on the product and package are understood. For example, did a validation study provide objective evidence that resterilization was acceptable?
If the firm's Quality System failed to recognize the process or product nonconformance(s) or take appropriate CAPA, this may be a major CAPA deficiency. Review the firm's equipment adjustment, maintenance and calibration records for the process. These activities may provide further insight into the cause of the nonconformances.
Examples of nonconformances and sterilization process failures the investigator may encounter include: Test Failures (e.g. Positive Biological Indicators, high EO residues, high bioburdens, out of specification endotoxin results); Parametric Failures (process failures such as unspecified dwell times, low pressure, low EO gas weights, loss of humidity, etc.); and Packaging Failures. Packaging Failures may be an indication of a sterilization process parameter problem (vacuum) or a packaging process problem (validation, sealer set up, etc.).
Important linkages to consider at this point include Corrective and Preventive Actions, Material Controls (820.90 Nonconforming product), and Facilities and Equipment Controls (820.72 Control of inspection, measuring, and test equipment).
4. If the sterilization process is software controlled, confirm that the software was validated.
If the sterilization process chosen is NOT controlled with software, proceed to Objective 5.
If the sterilization process is automated with software, review the software requirements document, software validation protocol, software validation activities, software change controls and software validation results to confirm that the software will meet user needs and its intended use. If multiple software driven systems are used in the sterilization process, challenge one based upon significance.
An important linkage to consider at this point is Material Controls (820.50 Purchasing Controls). For example, for software developed elsewhere, confirm that appropriate software and quality requirements were established and provided to the vendor and that purchasing data (and validation results) support that the requirements were met.
5. Verify that personnel have been appropriately qualified and trained to implement the sterilization process.
Using the sampling tables, select a number of training and qualification records for process operators and employees conducting Q.C. activities related to the sterilization process. Where a process is operated over more than one shift, training records from all shifts should be included within your review. Confirm that all employees are aware of the device defects that may occur as a result of improper performance of their assigned responsibilities. Confirm that employees conducting Q.C. inspections and tests are aware of the defects and errors that may be encountered while performing their assigned responsibilities.
An important linkage to consider at this point is Management Responsibility (820.25 Personnel).
NOTE: Information that must be reported with the Establishment Inspection Report (EIR) includes: 1. The identification of all sterilization processes used by the firm (e.g. Ethylene Oxide, Gamma irradiation, etc.); 2. The identification of the sterilization process covered; 3. The identification of any standard that the firm claims to follow for the process covered (if applicable); 4. The location of the sterilization sites; 5. The division of responsibilities for sterilization services (e.g. contract testing labs, sterilizer, finished device manufacturer, packaging, labeling etc.); 6. The SAL ; and, 7. whether or not parametric release is utilized.