

Aseptic Processing and Packaging for the Food Industry
Updated: 2005-07-14
GUIDE1 TO INSPECTIONS OF ASPECTIC PROCESSING AND PACKAGING FOR THE FOOD INDUSTRY
1This document is reference materials for investigators and other FDA personnel. The document does not bind FDA, and does not confer any rights, privileges, benefits, or immunities for or on any person(s).
This document is also available in

TABLE OF CONTENTS
Clean-up and Re-Sterilization After Process Deviation
Scheduled Process for Reprocessed Products
CONTAINER STERILIZING, FILLING AND CLOSING OPERATIONS
PAPERBOARD OR PLASITC CONTAINERS
THERMOFORM-FILLED-SEAL CONATINERS-PRE-STERILIZED BY HEAT OR CO-EXTRUSION
TABLE 1: ADVANTAGES AND DISADVANTAGES OF HEATING SYSTEMS
Figure # 3: Product-to-Product
Figure # 4: Tubular Heat Exchanger
Figure # 5: Scraped Surface Heat Exchanger
Figure # 6: Superheated Steam Metal Container System
Figure # 7: Webfed Paperboard System I
Figure # 8: Webfed Paperboard System II
Figure # 9: Preformed Cup System.........pg. # 20
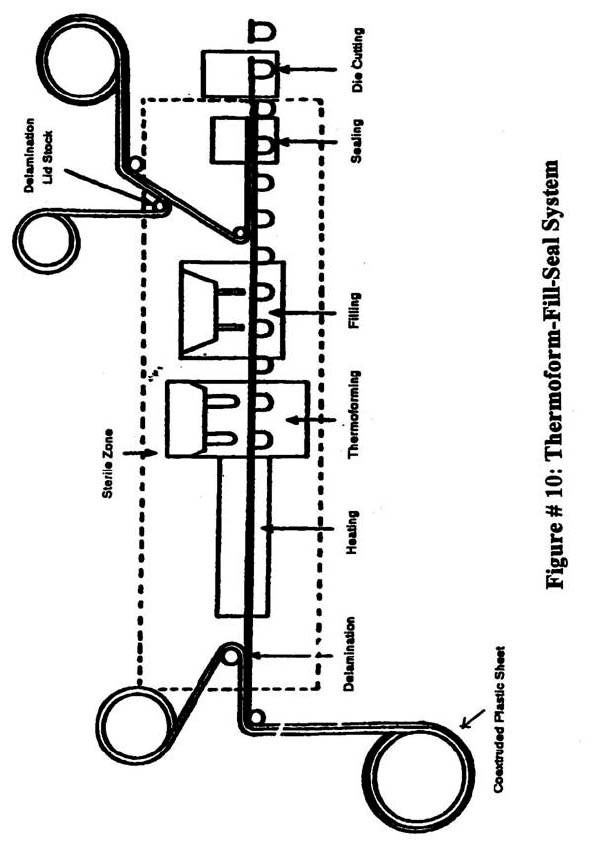
Figure #10: Thermoform-Fill-Seal System
INTRODUCTION
Inspections of aseptic processing and packaging systems for Low Acid Canned Food (LACF) are some of the most complex inspections of food manufacturing operations. The major difference between aseptic processing and the more "conventional" types of LACF processing is that a process authority(s) must establish a process that ensures commercial sterility not only of the product but also for:
- the product sterilization system (hold tube) and all equipment downstream from the holding tube including the filler;
- the packaging equipment; and
- the packaging material.
Documentation of production operations must be maintained by the firm showing that commercially sterile conditions are achieved and maintained in all these areas. Any breach of a scheduled process for the processing or packaging system means that the affected product must be destroyed, reprocessed or segregated and held for further evaluation. In addition, the processing and packaging system must be cleaned and re-sterilized before processing and/or packaging operations can resume.
Aseptic processing equipment sterilization procedures often use steam or hot water under pressure. Packaging equipment and packaging materials are sterilized with various medium or combination of mediums (i.e., saturated steam, superheated steam, hydrogen peroxide and heat and other treatments). Sterilization procedures are often validated by placing resistant microbial spores on adhesive strips at strategic locations in equipment or on container materials. Results of microbial validation studies are filed with CFSAN in support of scheduled process filings.
In addition to instructions and information provided in the Guide To Inspections Of Low Acid Canned Food Manufacturers (Parts 1, 2 and 3, hereafter referred to as the LACF Inspection Guide), direct attention to the following points when inspecting firms using aseptic processing and packaging. Before conducting the inspection, review the file jacket for the firm for previous establishment inspection reports (EIR's) and other pertinent information. Previous EIR's may provide a history of the installation of new, or modifications to, equipment and instrumentation. Review prior documentation dealing with incidents such as recalls, container integrity problems (involving leakers, swollen containers and visual external defects).
INSPECTION
Process Flow Chart
It is important to become thoroughly familiar with each step in the process, before attempting to evaluate the system for compliance with 21 CFR 108 and 113. This includes those components that are responsible for controlling critical elements in the process.
At the beginning of the inspection, obtain a diagram or blueprint of the entire processing and packaging system and conduct a walk-through review of the system, noting the various components on the diagram. In some cases, the firm may have a diagram or blueprint only of the product sterilization portion of the line, i.e., that portion from the raw product tank to the filler. If the diagram is only for a portion of the line, supplement this with your own diagram(s).
If a diagram or blueprint is not available, prepare a process flow diagram - from incoming raw materials to finished product warehouse storage. The critical control points - those points where lack of control could cause, allow, or contribute to a microbiological hazard in the final product - should be identified on the process flow diagram.
The firm should have written instructions for the operation of the product and packaging system, including pre-sterilization procedures to bring the product sterilizer (hold tube) and equipment downstream to the filler and the packaging system to commercial sterility prior to onset of product sterilization and/or packaging. Obtain copies of these procedures and submit with the diagram, blue print or process flow diagram as an exhibit to the EIR.
If the firm employs more than one aseptic processing system i.e., one product sterilization unit combined with one type of packaging unit (e.g., Dole, Tetra-Pak or Conofast), choose the system which appears to offer the greatest potential for contamination if the critical control points are not controlled.
Focus the inspection on one complete aseptic process and packaging system. If, after making an evaluation of the system you have selected, it is deemed necessary or advantageous to cover another during the same inspection, do so; but only if you can devote sufficient time to thoroughly evaluate a second system.
Scheduled Processes
Scheduled processes must be filed with FDA listing the critical factors necessary to reach a condition of commercial sterility for:
- the product,
- the "sterile zones" of the product sterilization system (hold tube and equipment downstream from the hold tube),
- the packaging system and,
- the packaging material.
The firm is required to list such information on Form FDA-2541c and its attachments. Become familiar with this form and the "Aseptic Packaging System Supplement" to the instructions for establishment registration and process filing for acidified and low-acid canned foods. Copies of each should be available in your district or they may be obtained from the LACF Registration Coordinator at (202) 205-5282.
Review and compare copies of the firm's current scheduled processes with those filed with FDA. Filed processes may have been obtained during a previous inspection; or they may be obtained using procedures outlined in the LACF Inspection Guide - Part 1. Supplemental information on pre-sterilization and sterility maintenance of processing, packaging equipment and sterilization of packaging material can be obtained from the Center for Food Safety and Applied Nutrition (HFS-617 - Regulatory Food Processing and Technology Branch).
Review the scheduled processes used by the firm to assure they have been recommended by a process authority (letter, standard operating procedures manual, transmittal, bulletin, etc.). Do not routinely request actual process establishment information unless instructed to do so by your district and by HFS-617(See the LACF Inspection Guide - Part 1). Compare the critical factors in the filed scheduled process to make sure they correspond to those in the transmittal from the process authority. Compare the filed process with the written documentation from the process authority prior to the walk-through for a more efficient evaluation of the critical components in the line.
Review the section in the supplement entitled "Required Supplemental Information For Aseptic Packaging Systems", which details information concerning the procedures necessary for bringing the processing and the packaging unit to a condition of commercial sterility and maintaining commercial sterility throughout processing operations.
PROCESSING
Product Heating Systems
There are three basic types of product heating: direct, indirect and ohmic. Each type will be discussed separately. There are advantages and disadvantages to using each type of heating system, the advantages and disadvantages are listed in Table I on page 14.
Direct heating systems -involve having steam condense into the product. This can be done in two ways:
- Steam injection (Figure 1): where steam is injected directly into product flowing through an injection chamber.
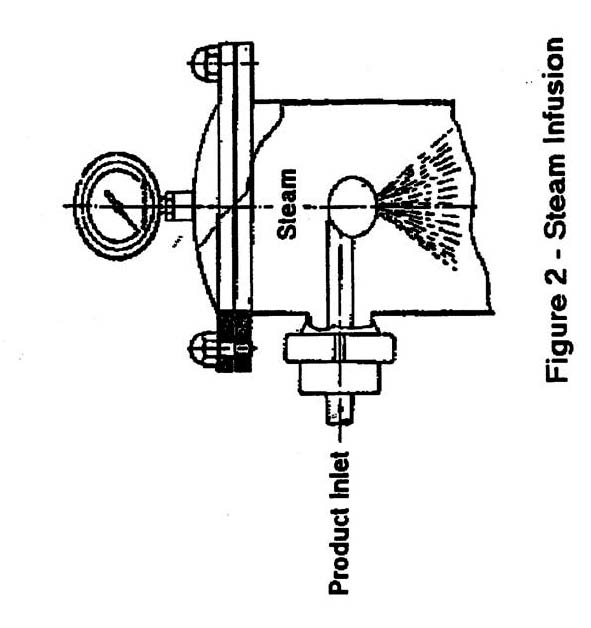
- Steam infusion (Figure 2): where the product is sprayed into a large pressurized steam chamber and is sterilized when falling as film or droplets through the chamber. In most cases, a flash chamber is used after the hold tube to evaporate added water, which results in a rapid cooling of the commercially sterilized product.
Indirect Heating Systems –Involve the use of equipment to exchange heat between the surface that is heated, and the product. There are three major way the indirect heat is used in aseptic processing. They are:
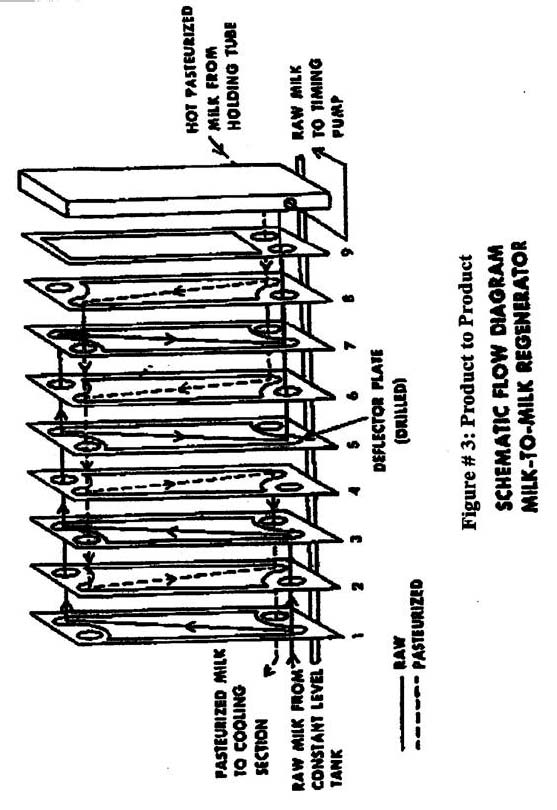
- Plate heat exchangers -(Figure 3), Where the plates in the system serve as a heat transfer surface and barrier with circulating hot water (for pre-heater) on one side and product on the other. This system is similar to those used in the pasteurized milk industry, are used most often for homogeneous liquids such as milk and other dairy products.
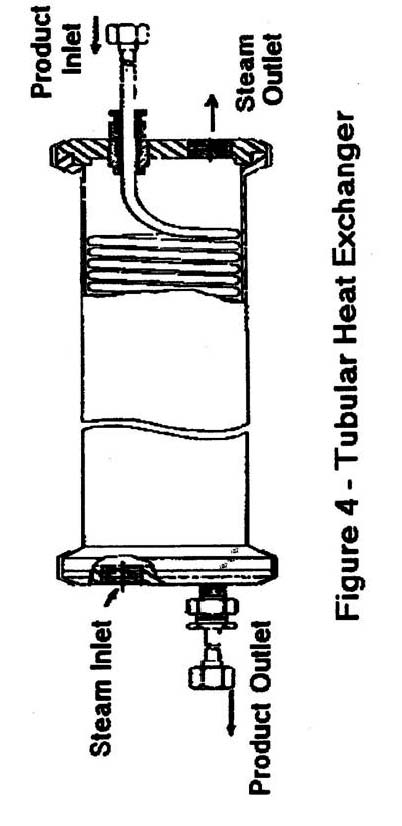
- Tubular heat exchangers-(Figure 4), generally employ concentric tubes as the barrier/ heat exchange surface. Product flows through the inner tube of two-tube systems and the middle tube in three-tube systems, with the heating medium flowing in the opposite direction through the other tubes. With shell-in tube heat exchangers , as shown in figure 4, the tube may be coiled inside a large shell, with product also flowing through the tube in a direction opposite to the flow of heating medium. As with plate heat exchangers, generally homogeneous products such as milk are normally processed in these systems.
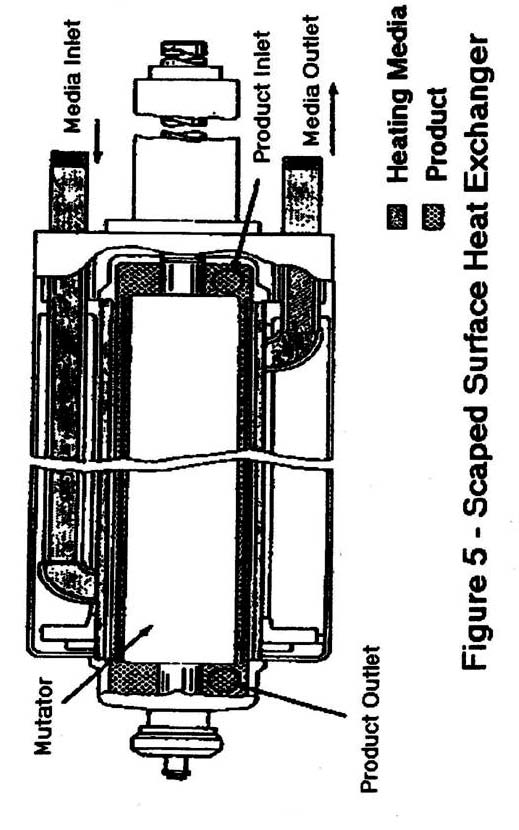
- Scraped-surface heat exchangers -(Figure 5), consists of a mutator shaft with scraper blades, generally concentrically located within a jacketed, insulated heat exchanger tube. Product is pushed against the inner heat exchange/barrier wall by the force of a pump, which transports product through the heater. The blades, which have a slight degree of independent movement, then "scrape" product build-up off of the heat exchange surface. The heating medium is circulating water or steam which flows on the opposite side of the inner heat exchange/barrier wall.
These heat exchange systems are normally used for processing viscous products, such as puddings, or products containing particulates such as certain soups.
Ohmic heating - is a relatively new method of product heating where an electrical current is passed through a suitable conducting product causing product heating. The system operates under continuous flow conditions with the product passing over electrodes in one or more heating tubes, followed by product cooling in scraped surface, tube in shell or plate heat exchangers. The conductivity and electrical resistance of the product influences the heating rate. Because of this, product formulation becomes critical to the process. Food products, which are not good conductors of electrical current, are not good candidates for ohmic heating.
Equipment and Controls
Raw Materials and Formulation
Describe the firm's method for ensuring the microbiological quality of its raw materials.
Determine whether any formulation changes (e.g., changing starch types or amounts) might adversely affect the adequacy of the thermal process.
Determine how the firm controls the formulation and batching of product to insure that the product meets the desired characteristics.
If the product contains particulates in its formulation (tapioca pudding, certain sauces and soups, etc.), review the scheduled process transmittal from the processing authority in order to determine the critical factors associated with the particulates. Document that the firm's procedures are sufficient to meet the appropriate levels for these critical factors. For example, if particle size is a critical factor according to the processing authority, determine how the firm insures that the size of the particulates used is at or below the size specified or if a rehydration period is required for dry particulates. Document the firm's procedures to meet the requirements specified by the processing authority.
Metering(Timing) Pump
Aseptic processes are based on a continuous flow of product through a holding tube. This continuous flow relies on pumps, and as such, these pumps are critical in the design of the system. 21CFR 113.40 (g)(i)(f) states "A metering pump shall be located upstream from the holding tube and shall be operated to maintain the required rate of product flow". A positive displacement pump is used as the metering (sometimes called the timing) pump because they are less sensitive to pressure drops and slippage than centrifugal pumps. The product characteristics may determine the type of positive displacement pump used. When the pressure drop in the system is low (less than 150 lbs.) and the product contains only small particles or is homogenous, a rotary positive displacement pump may be used. At higher-pressure drops and for large particulates, a reciprocating piston pump is normally the pump of choice.
Metering(Timing) pumps may be variable speed or fixed-rate. In the latter, the pumping rate cannot be changed without dismantling the pump. If the pump is a variable speed device (e.g., has a Reeves-type drive), a means of preventing unauthorized speed changes must be provided. This can be a lock on the device or a notice from management posted on or near it, giving suitable warning.
Flow Meters
Some newer systems may use a flow meter to control the flow of product through the system. The flow meter may be used in conjunction with a fixed rate pump and a flow control valve or with a variable speed pump controlled by the flow meter. When these flow control systems are used it is extremely important to determine how the flow control system operates, the procedures used to validate the flow rate, and how the system is maintained.
Product flow rate through the hold tube affects the residence time of the individual element in the hold tube. Each fluid element may receive a different degree of sterility, depending on the length of time that particular element spends in the hold tube (residence time). The design of the system, pumping rates and product characteristics can effect the flow rate through the heating and holding portions of the system. This is why product formulation is critical.
The residence time of the fastest moving element is determined and calculated by the processing authority for the product being heat processed. Then the processor documents that the flow rates and flow characteristics of the product are not different from those established by the process authority. The specified product flow rate should be monitored or verified by the processor as a routine part of the system operation. One method of doing this is by correlating the flow rate under no load conditions with the pump speed. By counting pump strokes per set time period, or by equipping the pump with a recording tachometer, an indirect record of product flow rate can be documented. The efficiency of some pumps may be affected by viscosity of the product and the absence of pressure or backpressure in the system. Thus pumping rates established with water may not reflect a true flow rate for the food product. Various types of flow measuring devices have been developed which indirectly provide an indication of product flow. The use of a flow meter to indicate product flow rate should be validated by the firm and documentation should be available which supports the use of the flow meter as an accurate indication of actual product flow. Physical measurement of product flow (e.g. 3 gal per minute, number of containers per set time interval) may be an acceptable method to determine product flow rates. Sampling sites and product temperature must be specified, as product temperature may have an effect on product volume. If product is going directly to a filling line, product fill rates can be determined over a set time period and correlated to product flow rates. Means do exist where chemical or radiological tracers are injected into the product stream to measure product flow. However these methods are normally not used on a routine daily basis to verify product flow rates.
Document the firm’s validation procedures for insuring that the pumping rate determined by the processing authority is met by the system.
Sterilizer (Hold Tube)
Temperature Indicating Device
Sterilizers or hold tubes must be equipped with at least one TID’s: During the inspection, check that the device complies with the specifications listed in 21 CFR 113.40(g)(l)(i)(a). which gives the parameters of the TID and how often it is checked for accuracy.
If the system is equipped with only one temperature indicating device, the probe for this device is normally located in the vicinity of the temperature recording device. Mercury-in-glass thermometers and other temperature indicating devices are the reference instrument and, as the regulations state, “shall be tested for accuracy against a known standard upon installation and at least once a year thereafter.” The regulations do not require maintenance of calibration records, however, as the reference instrument for indicating the processing temperature it is important that the firm be able to document that required tests for accuracy were accomplished. If possible obtain copies of the records of testing for accuracy as well as copies of the method used, and the name of the firm or individuals performing the tests.
Temperature Recording Device
21 CFR 113.40(g)(l)(i)(b), states in part: “The temperature recording device shall be installed in the product flow at the holding-tube outlet between the holding tube and the inlet to the cooler.” It goes on to say: “The temperature chart shall be adjusted to agree as nearly as possible with, but to be in no event higher than, a known accurate mercury-in-glass thermometer. “
The firm must also have a means to prevent unauthorized changes in adjustment of the recording device.
Temperature Recorder-Controller
21 CFR 113.40(g)(l)(i)(c). Describes where an accurate temperature recorder controller shall be located an the specifications of the recording chart.
Describe the operation of the recorder-controller and report the name of the manufacturer. It goes on to say that if it is air-actuated, the firm needs to assure a supply of clean dry air to the controller. Check to see if there is filter on the air line and if so, how often is it changed or monitored to assure the quality of air to the controller.
In some instances, a firm may have a recorder-controller with two or more pens, one marking the recorder-controller temperature at the exit end of the final heater; and one recording the temperature at the exit end of the holding tube. Describe how the firm adjusts the recorder controller, how often, and what reference instrument is used to adjust the recorder controller. The pens are adjusted by reading the indicating thermometer and using a thumb wheel on the pen on making adjustments inherent in the particular design.
Product to Product Regenerators
21 CFR part 113.40(g)(1)(i)(d), states: “When a product-to-product regenerator is used to heat the cold unsterilized product… it shall be designed, operated and controlled so that the pressure of the sterilized product in the regenerator is greater than the pressure of any unsterilized product in the regenerator to ensure that any leakage in the regenerator is from the sterilized product into the unsterilized product.”
Differential pressure recorder-controller
21 CFR 113.40(g)(1)(i)(e). Is the means of controlling pressure in a product-to-product regenerator. The device must comply with the specification listed in the regulation and the nature of the control action taken by the device in the event of improper pressures. It is necessary for one pressure sensor to be located in the sterilized product regenerator outlet (point of lowest pressure) and one in the non-sterilized product regenerator inlet (point of highest pressure).
During the inspection, note were the sensors are located and document then on the process flow diagram. Also, the controller must “be tested for accuracy against a known accurate pressure indicator upon installation and at least every three months of operation thereafter, or more frequently, if necessary... “ This part of the regulation does not address a record-keeping requirement or recommendation relative to this testing schedule, however, it is important that the firm keep such records. Review copies of the records of testing as well as a copy of the methodology used and determine the name of the firm or individuals performing the tests. This information should be included in the EIR.
Product Holding Tube
21 CFR 113.40(g)(1)(i)(f). The regulation states that the product sterilizing holding tube must be designed to give continuos holding of every particle of food for at least the minimum holding time specified in the scheduled process.
To assure this, the tube must be sloped upward at least 0.25 inches per foot. Pitch of hold tube can be determine with a T square or by using a line level. Verify that the holding tube diameter and length conforms to that listed in the filed scheduled process and that the slope is adequate. If the holding tube is capable of being dismantled (for cleaning, repairs, etc.), record in the EIR how the firm assures that when reassembled, it conforms to the scheduled process parameters.
Also, the holding tube must be designed so that no portion of the tube between the product inlet and the product outlet can be heated. Hold tubes may be insulated to protect the hold tube from external extreme temperatures. This is acceptable as long as no external heat source is applied to the hold tube.
Flow diversion system-
21 CFR 113.40(g)(1)(i)(h). Describe in detail the firm's method for diverting non-sterile product flow away from the filler or aseptic surge tank, including any documentation from a processing authority that may list specific recommendations for the design and operation of the system. Some firms may elect to install a flow-diversion system. The regulation describes where the device should be located but does not require the placement of the device in the line. If it has an automatic flow diversion device or system, document the variables (e.g., loss of temperature, loss of pressure in a product-to-product regenerator, etc.) that will activate it to divert flow. Document what system is in place to notify the operator to divert manually operated systems. Record how diversion incidents are recorded, including corrective action and disposition of diverted product. Verify that the first divert drain is sterilized following each use and that a gravity-drain flow diversion device is not used.
Equipment downstream from the holding tube
21 CFR 113.40(g)(1)(i)(i). Entry of microorganisms into the product can happen at product coolers, aseptic surge tanks, flow diversion valves, homogenizers, aseptic pumps or any other equipment that is downstream from the holding tube. Rotating or reciprocating shafts and valve stems should be equipped with steam seals or other effective barriers at the potential access points. The firm needs to monitor the performance of these seals or barriers for proper function during operations.
Aseptic Surge Tank - are sometimes employed by a firm as a means to temporarily store sterile product. This is done to provide a continuous supply of product to the filler or to divert sterile product in the event of a stoppage of the packaging machine. Surge tanks are sterilized before start-up of product flow with steam or water up to any air filter in the line or up to the filler valve. Generally, aseptic surge tanks must be vented, in a manner similar to still retorts, to ensure there are no remaining air pockets, which would prevent certain areas within the surge tank from reaching sterilization temperature.
Typically, a processing authority establishes this “venting” or air purge schedule, and the firm has documentation from this authority, specifying the sterilization procedure. During the inspection review the firm’s data to make sure the purge schedule is listed. Sterile air over-pressure must be maintained on aseptic surge tanks to ensure proper operation (i.e., product flow to the filler). Sterile air or gas is produced by incineration and/or filtration. Determine how the firm monitors sterile air or gas over-pressure and the method of achieving sterility. With incineration, a thermocouple monitoring system is probably the easiest means. If a sterile filter is used, determine the specifications of the filter, filter location and number of filters. Determine if the firm changes the filter at intervals recommended by the manufacturer or process authority for their method of use. Filter changes should be documented on the processing records. Determine whether the firm has taken into account any possible adverse effects that may affect the working life of the filter, such as, repeated contact with incinerated air.
If a filtration system is used and the downstream side is sterilized with steam during the vent or purge cycle, determine whether the process authority or the manufacturer took into account the effects of steam on the filter. The firm should have a procedure to determine the integrity of filters.
There are several commercial methods for testing filter integrity, but basically the firm should use the method recommended by the filter supplier or their process authority. Loss of filter integrity is a process deviation and places the commercial sterility of all product produced in question.
Gases, such as sterile nitrogen or carbon dioxide - either singly or in combination - may be used to provide overpressure and create a sterile barrier. Determine the firm's procedure for ensuring the sterility of these gases and any filters used to filter the sterile gases including lines/piping downstream to the point where the gases are delivered to the aseptic system.
Backpressure - Backpressure valves or orifices may be used in aseptic systems to assure that pressure in the system prevents flashing of the product in the hold tube. Flashing in the holding tube may cause an increase in velocity of the product, thereby reducing the residence time specified in the schedule process. Determine how the firm monitors the proper operation of the backpressure valve(s). For examples, in direct heating systems (e.g., steam injection or infusion), the added water from the condensed steam must be removed for standardized products such as milk. This is normally done in a sterile "flash" or expansion chamber. A firm must have a back pressure valve to separate the holding tube from the flash chamber in order to prevent "flashing" (i.e., water vapor expanding as steam) from taking place in the holding tube.
Control system- Product heating and sterilizing systems run the entire spectrum, from manually operated systems to computer-controlled highly automated systems. For the manually operated systems, review of the production logs and recording charts by management represent the principal method of verifying that the product received the scheduled process. For highly automated systems, there are controls that, if operating properly, will automatically preclude the packaging of non-sterile product into sterile containers. Consequently, routine challenge and calibration of the automatic controls represent an additional method of verifying that the product received the scheduled process. During the inspection, obtain from the firm a copy of the most recent challenge and calibration record for the automatic controls. Included should be the methodology employed, the frequency of testing, and the individuals who conducted the tests. Computerized control systems must be validated upon installation to insure that they will operate as designed.
OPERATION
21 CFR 113.40 (g)(l)(i)(ii)(a): The firm must follow its filed scheduled process for bringing the equipment to a condition of commercial sterility (i.e., as listed in the "Required Supplemental Information for Aseptic Packaging Systems") prior to "switching-over" to product sterilization. Determine if the temperature sensor, which is monitoring the equipment sterilization temperature, is located at the defined coldest point in the line, downstream from the hold tube. This sensor is usually located at a point beyond the valve, which interfaces the hold tube with the filling equipment. If, for example, the firm is using the temperature indicating device (e.g., a mercury-in-glass thermometer) at the exit end of the holding tube to indicate equipment sterilization, document how the firm assures that the equipment downstream of the holding tube reaches the proper temperature.
And, determine how the firm assures a proper switchover from water to product without causing a process deviation to occur in either the equipment sterilization or product sterilization cycle. For example, the sterilization temperature for bringing the equipment to a condition of commercial sterility may be several degrees F more - or less - than that which is scheduled for the product.
21 CFR 113.40 (g)(l)(i)(ii)(e). Monitoring records for aseptic processing should as appropriate include readings for the following
- Temperature indicating device(s) at holding tube outlet.
- Temperature recorder at holding tube outlet.
- Temperature recorder-controller at final heater outlet.
- Differential pressure recorder-controller, if a product-to-product regenerator is used.
- Product flow rate (in gallons per minute, cans per minute, etc.).
- Aseptic surge tank sterile air overpressure or other protective means.
- Proper performance of steam seals.
- The sterilization of equipment or "pre-sterilization" cycle. The records should indicate when the equipment is in the pre-sterilization cycle, when flow diversion occurs and when product is flowing through the system.
Following is a list of some possible process deviations:
- Temperature drop in the holding tube.
- Loss of differential pressure in product-to-product regenerator.
- Loss of sterile air or gas pressure or other protection level in the aseptic surge tank.
- Loss of sterility of air or gas supplies to sterile zones.
- Critical factors in the scheduled process outside specifications.
- Increasing the speed of the variable speed-metering pump.
If a process deviation occurs and potentially non-sterile product is filled into a container, the firm must perform corrective action on the affected product. This could include reprocessing or destroying the product or having the process evaluated by a processing authority. During the inspection, review all process deviations and, if the firm chose to have a deviation evaluated by a process authority, collect those records and responses and submit them as an exhibit to the EIR.
Clean-Up and Re-sterilization After Process Deviations
The firm should have written procedures for ensuring effective clean up and re-sterilization of the sterile product portion of the line after a process deviation. Determine the re-sterilization cycle for the equipment. If it is different, determine if a process authority recommended the re-sterilization cycle, and if the firm has a letter or other form of documentation establishing the parameters of the re-sterilization cycle.
When reviewing process deviation records, make sure they document:
- Clean up of the system following a process deviation.
- Return of product sterilizer and all downstream equipment to a condition of commercial sterility.
- Disposition of any suspect product filled into containers.
If the firm does not keep the appropriate records, this should be an item for discussion on the FD483.
Scheduled Process for Reprocessed Products
If a firm decides to reprocess product that was involved in a deviation, several factors need to be taken into account, as not all products will flow the same. If the original product has turbulent flow characteristics, the product could exhibit laminar flow characteristics after the first process. This is especially true of products containing starch or other binders. Also, factors such as reprocessing the affected lots separately or together; or blended with new product can affect the process. During the inspection, determine if the firm has considered all factors that might affect reprocessing have been taken into account.
There are a variety of aseptic filling and packaging systems currently used in the United States for acid and low-acid foods. Generally, these may be described by inclusion in one of six categories:
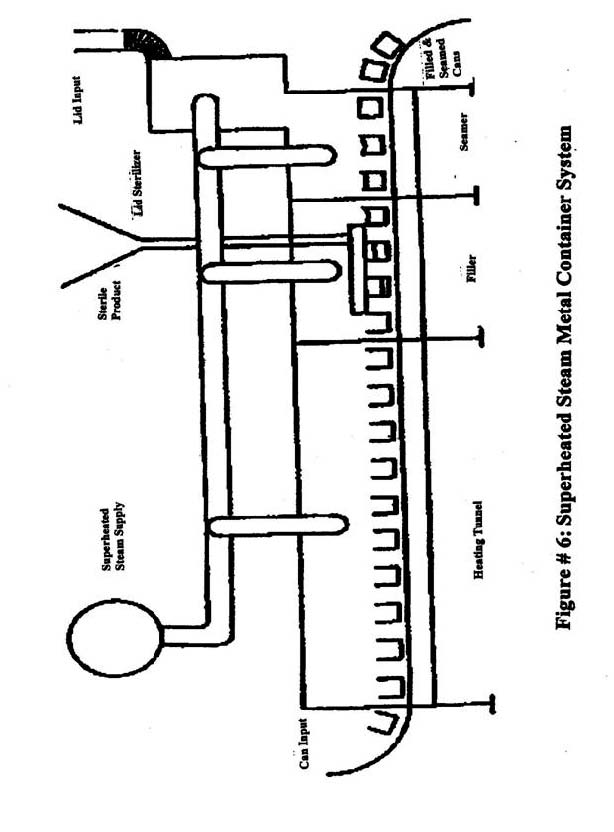
- Metal containers and closures: Figure 6 shows a system where containers are sterilized and filled using superheated steam as the sterilizing medium. (e.g., the superheated steam system).
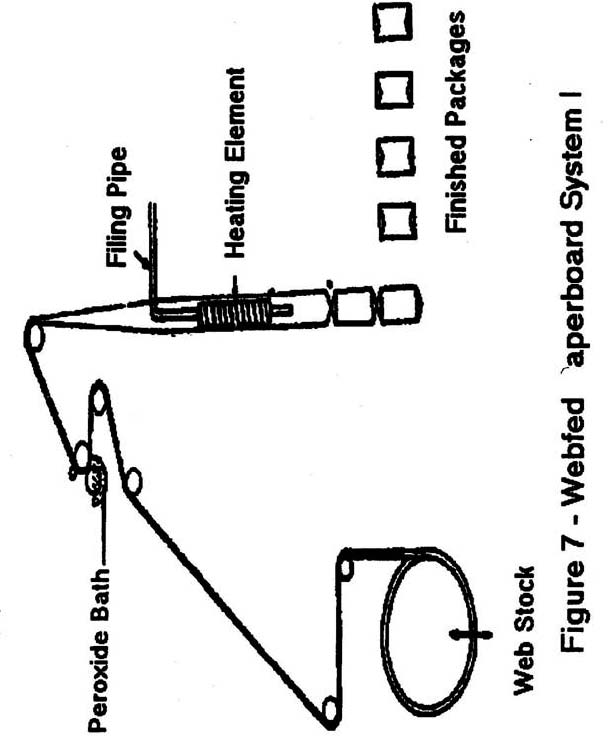
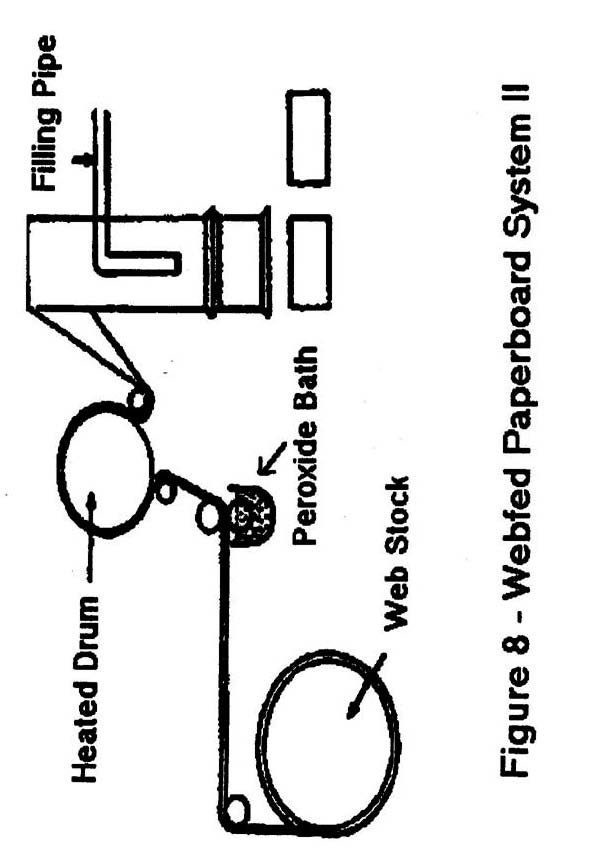
- Webfed paperboard: Figure 7 and 8 are sterilized using hydrogen peroxide and heat.
- Preformed or partially formed paperboard is sterilized using hydrogen peroxide and heat.
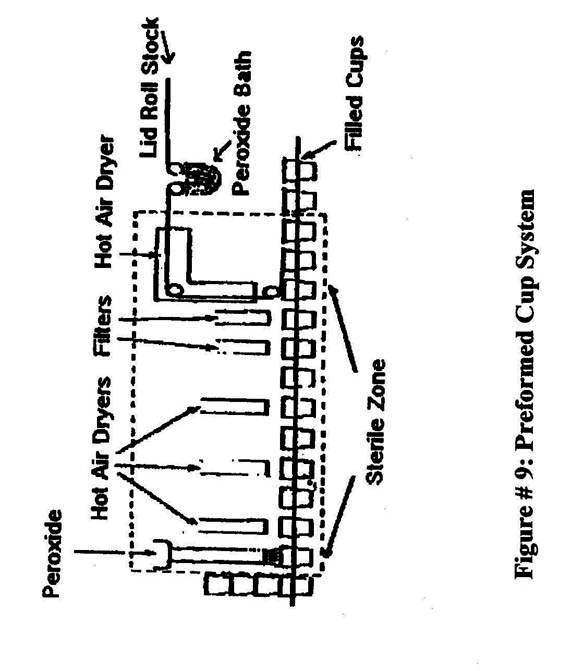
- Preformed plastic cups: Figure 9 are also sterilized using hydrogen peroxide and heat.
- Thermoform-fill-seal: Figure 10 shows a system which uses hydrogen peroxide and heat or the heat of co-extrusion to sterilize.
- Bag-in-Box system uses containers pre-sterilized by gamma irradiation.
The firm must file a scheduled process for its packaging system, including supplemental information regarding the critical factors involved in bringing the system to a condition of commercial sterility prior to start-up. This is done on Form FDA 2541c - "Food Canning Establishment Process Filing For Aseptic Packaging Systems." Also, the firm must have on file, a letter or other documentation from a process authority, which supports the filed scheduled process. Obtain a copy of the document, compare it to the filed scheduled process and become thoroughly familiar with the critical factors involved. A deviation from any of these specified critical factors constitutes a process deviation, which must be handled in accordance with 21 CFR 113.89.
CONTAINER STERILIZING, FILLING AND CLOSING OPERATIONS
Metal Containers and Closures
Recording devices- In order to demonstrate that the required sterilization is accomplished, firms use automatic recording devices. During the inspection it is important to document the number, location, and type of sensors used. In a steam sterilization system, such as the Dole unit, the basic components of the system are:
- container sterilizing section,
- filling section,
- cover or lid sterilizing unit, and
- container closing section.
You should also know what critical factors are being monitored, e.g., temperature, sterilization media flow rate, etc...And determine if they are being recorded accurately. After identifying where the recording devices are, check to make sure equipment correspond in number and location to those on the filed scheduled process.
The firm also needs to assure that their recording devices are accurate.
If indicating-type thermometers are used, they must agree with the recording thermometers. For TID’s(Temperature Indicating Devices), determine if, how and when the firms
calibrate them and if calibration is accomplished at the scheduled process conditions.
Obtain a copy of the last calibration, the methodology used, and who conducted the test. It is important that monitoring equipment be calibrated in the range of operation (e.g., if the temperature is 200°C (400°F) for hot air used for container sterilization, then the monitoring equipment should be calibrated near that temperature).
Sterile Water- In aseptic systems using metal containers and closures, if cold sterile water is directed against the bottom of the containers after filling (or on the lids prior to closing), determine the firm's controls for ensuring the sterility of the water on a continual basis. If non-sterile water entered the filling area, this would constitute a process deviation.
Timing Method- Describe the firm's controls for ensuring the proper residence time of the containers and lids in the sterilizing medium. Check the container/closure flow rate with a calibrated stopwatch. If the firm uses an automatic device to monitor container/closure flow rates, determine how does the firm assure these devices are accurate. Describe the method for preventing unauthorized speed changes.
For the Dole can sterilizer, it is crucial that the sterilizer be at full speed at all times since any gaps in container flow through the sterilizer can result in some cans being retained for less than the scheduled time. This is because the conveyor or chain through the sterilizer may attempt to "catch-up" the trailing cans to the leading cans where a gap in containers occur.
Start-Up- The firm must follow their filed scheduled process for bringing the equipment to a condition of commercial sterility. A lack of proper time/temperature sterilization of the equipment is a process deviation and must be handled in accordance with 21 CFR 113.89. The thermocouple indicating sterilization temperature for the equipment will probably be the same one used to indicate the temperature during operations, and should be located in the most-difficult-to-sterilize area.
Suitability of containers and closures for sterilization-
Determine how the firm assures that containers and covers are clean and dry prior to entering the steam chambers. Wet containers or covers cause a condensation of steam at the surface of the container. As sterilization takes place under atmospheric conditions, this means that the temperature of the can bodies and/or covers would rise no higher than 100°C (212°F), whereas the temperatures employed in the superheated steam unit are designed to give can temperatures of approximately 215.6-218.3°C (420-425°F) and cover temperatures of approximately 210-212.8°C (410-415°F).
Loss of temperature- During start-up, failure to achieve the time-temperature listed in the scheduled process for containers, closures and equipment is a process deviation and must be handled in accordance with 21 CFR 113.89.
In the event there is a loss of temperature during filling, determine what corrective action the firm takes. The corrective action should include things such as, automatically or manually stopping the line, diverting the product, and fixing the problem. If product was filled into containers, part of the corrective action would be to make sure that affected product is segregated.
Decreased residence time- If there is a decrease in residence time, determine if there is an alarm system or automatic stop of the line. If not, determine if the product is diverted or if filled containers are properly segregated. The firm also has to assure that the unit is properly re-sterilized after the deviation.
Loss of sterility in cooling water- Determine what the possibility is that there would be a loss of sterility in the cooling water and what the firm’s procedures are for recognizing the deviation and what corrective action would be taken.
Clearing container jams -If an operator has to enter the sterile zone (e.g., the seamer) with tools to clear can jams; determine the firm’s procedures to re-sterilize the zone.
PAPERBOARD OR PLASTIC CONTAINERS
(Webfed, Pre-formed or Thermoformed are filled/sealed using Hydrogen Peroxide as a Sterilizing Medium)
Equipment and Controls- Describe in detail, the firm's procedure for monitoring the following (if they are filed as critical factors for the scheduled process):
- Peroxide consumption rate
- Peroxide concentration
- Peroxide level (if immersion method used) or deposition (if roller applicator or fogger is used)
- Temperature of warming air used to transport chemical sterilants
- Air or heating element temperature (for removal of H2O2 and completion of sterilization)
Note: Heat is generally applied by one of four primary methods:- A tube heater, located in the center of webfed paperboard as it is being formed into a tube.
- A horizontally placed heating element located above containers into which H2O2 has been sprayed.
- Air knives that blow hot sterile air against webfed paperboard or plastic after emergence from an H2O2 immersion tank and prior to forming.
- Water-heated stainless steel drum.
- Sterile air temperature (for incinerated air, subsequently cooled and used to provide over-pressure in a sterile zone)
- Sterile air filters
- Sterile air over-pressure
- Gas flush - nitrogen or other sterile gases used to flush equipment or container headspace must be sterilized and maintained in a sterile condition. Determine how the firm assures that the sterility of the gas is not compromised. Determine the maintenance schedule for filters used for sterile gases.
Determine if the sensors for monitoring the above factors are located to provide assurance that the factor is being monitored at its coldest or weakest point. Also, find out if the firm maintains nozzles that are used to spray chemical sterilants, or if pumps such as peristaltic pumps, are used to control sterilant spray volumes.
Operations
Start-Up- The firm must follow their filed scheduled process with respect to bringing the equipment to a condition of commercial sterility prior to filling. Firms will generally use a combination of steam or hot water (for the filling apparatus) and H2O2 mist or spray for the sterile forming (if necessary), filling and closing or sealing areas, collectively referred to often as the "sterile tunnel" or "sterile zone". Warming air to transport fog sterilants and drying air temperatures are often critical factors because they contribute to sterilization.
Packaging Materials, Handling Procedures-Determine the firm’s procedures for assuring high microbiological quality of packaging material received and used.
Process Deviations- Many of these packaging systems or units are equipped with controls which, if functioning properly, will automatically stop the machine and preclude the packaging of sterile product into insufficiently sterilized containers. Of critical importance is a determination that these controls or "guards" will operate as designed (i.e., in the event of a failure to meet a factor specified as critical to the scheduled process, the machine will, in fact, shut down automatically). Determine:
- Who calibrates or checks the automatic controls or guards for proper operation, and the frequency of these checks and obtain a copy of the last calibration methodology and results.
- How the firm challenges the control system and if possible, obtain a copy of the procedure as well as a copy of the most recent results.
Although these systems usually are operated in an automatic mode, most, if not all, appear to be equipped with a capability for a manual override of the automatic controls. Determine under what circumstances the machine would be operated in a manual mode, if product would be packed in this mode, and who has the authority to order such operation. Also determine if inadvertent manual operation be detected during a routine review of processing records.
With respect to any controls or guards which are not automatically controlled, and which are critical to the scheduled process, determine how a process deviation would be detected and how would such a situation be handled by the firm?
H2O2 residual testing
Describe the firm's procedure for testing for H2O2 residual on the packaging material. Is the residual level in compliance with 21 CFR Part 178.1005(d)?
THERMOFORM-FILLED-SEAL CONTAINERS-PRE-STERILIZED BY HEAT OR CO-EXTRUSION
Equipment and Controls - Describe the firm's procedure for monitoring the following (if specified as critical to the scheduled process):
- Pre-Sterilization (bringing equipment to condition of commercial sterility prior to thermoforming and filling)
- Surface temperatures in various components of the sterile zone.
- Hold time after temperatures have reached that specified in the scheduled process.
- Overriding air pressure in the sterile zone.
- Filters for sterile air providing overriding air pressure during processing operations must be changed after a specified number of uses because they are in contact with incinerated air during the pre-sterilization cycle. Determine how often filters are changed to comply with that specified in the scheduled process and how the firm documents this.
Describe measures the firm employs to ensure protection of the sterile inner layer of the cup and lid material as it is received and used. Describe procedures for splice sterilization of cup and lidstock material rolls.
Operation - Determine the firm's procedure for ensuring that equipment is brought to a condition of commercial sterility, and that exposure of the sterile inner layer to the sterile zone at the beginning of the pre-sterilization cycle is performed in such a manner as to maintain the sterility of both the packaging material and the sterile form-fill-seal area (sterile tunnel).
Process deviations - Obtain the same type of information as previously discussed under process deviations involving systems using chemical sterilizers (e.g., H2O2).
BAG IN BOX PACKAGE SYSTEMS
Several manufacturing firms offer large bulk bags capable of holding several hundred gallons of product for aseptic filling. These bags are normally pre-sterilized with radiation. The bags are stabilized by an outer carton during filling and shipping. The outer carton may be reused several times, however, the bags and filler valves are used only once. Each bag is equipped with a fitment or valve which, when matched with the correct filler will allow for aseptic filling and emptying of the bag. Depending upon the system the filler nozzle may be sterilized by using chemicals (i.e., hydrogen peroxide) or steam.
Equipment and Controls - Describe in detail the procedures for monitoring the following:
- Operation of the fitment.
- If steam is used to sterilize the fitment, how the sterilization process is monitored.
- If chemicals are used to sterilize the fitment, how the chemical concentration is monitored.
- What procedures the firm has in place to assure that sterile packaging materials are received and maintained sterile.
Note: Procedures used to sterilize the fitment are specified by a process authority.
RECORDS
Observations and measurements of operating conditions or factors critical to the scheduled process must be made and recorded at intervals of sufficient frequency to ensure the product is being maintained in a condition of commercial sterility. These measurements should be made at intervals not to exceed one hour. Describe the firm's record-keeping procedures for monitoring critical control points on the packaging systems or units. On initial inspections collect blank copies of record forms used and describe the type of information recorded.
CONTAINER CLOSURE EVALUATION
Describe in detail the firm's container closure evaluation system. For metal containers and closures, check for compliance with 21 CFR 113.60(a) & (a)(1). Determine the source and obtain copies of any container closure guidelines.
POST-PROCESS HANDLING
Describe the firm's post-process handling procedures. Check for compliance with specifications established by the container manufacturer. Describe in detail any deviations from container supplier recommendations for post-process handling.
TRAINING
Describe the firm's training program for operators of the product and package sterilization systems or units. The firm should maintain a documented training program for operators of sterilization and packaging systems. Determine if the equipment manufacturers offer additional technical support.
MAINTENANCE
Evaluate the firm's maintenance procedures packaging systems. Pay particular attention to that portion of the product sterilization system downstream of the holding tube. For example, plate-to-plate heat exchangers are susceptible to pin holing, flex cracks and gasket leaks. This would be a critical maintenance area for such a heat exchanger (such as a product-to-product regenerator or product cooler) located downstream of the holding tube. Proper maintenance of in-line static seals or gaskets in system piping downstream of the holding tube, particularly from the exit end of the final cooler to the filler is also critical. The firm should maintain a documented maintenance program for all equipment. Maintenance should be performed on at least the minimum schedule recommended by the equipment manufacturer.
SAMPLE COLLECTION
See IOM Sample Schedule Chart 2 for guidance.
Incubation Tests. Incubation is not a mandatory requirement of the regulations. When performed by a firm determine:
- if containers are statistically sampled.
- how many containers are incubated.
- time and temperature of incubation.
- firm specifications for acceptability of lot.
- if spoilage is detected by firms, do they perform spoilage diagnosis to determine cause. Specify method and describe lot disposition procedures.
Procedures for the evaluation of various containers used for packing aseptic LACF are contained in the LACF Inspection Guide Part 3.
TABLE I: ADVANTAGES AND DISTADVANTAGES :
DIRECT HEATING
The advantages are:
- Rapid heating and cooling times resulting in less organoleptic damage to the product during heating.
- Less fouling or "burn-on" of product in the heater.
The disadvantages are:
- Because of the large volumes of steam that must be condensed, direct heating systems may be more difficult to control.
- The addition of water (from the condensation of the steam) increases the product volume by approximately 1% per 10°F temperature increase above initial product temperature as it enters the product sterilizer. This increase in product volume must be compensated for by the process authority establishing the thermal processes if flow rate is controlled prior to direct heating. If such, initial temperature must be controlled and recorded (volume increases with temperature). For these systems flow rate would not need compensation for volume increase.
- Steam used in direct heating of the product must be of culinary quality (appropriate for food contact). Culinary steam should be produced under conditions that meet safe boiler water requirements. Verify that the boiler water treatment compounds labeling meet the requirements of 21 CFR 173.310 by checking labeling of inventory or by examining letters of guarantee.
INDIRECT HEATING
The advantages are:
- You can control temperatures of the food to be heated.
- You can process viscous products (i.e., purees, puddings and shake bases) without burning the food.
- Energy conservation (e.g., using sterilized product to heat unsterilized product and thereby cooling the sterilized product).
The disadvantages are:
- Particle shear can occur.
OHMIC HEATING
The advantages are:
- Rapid heating
- Can process foods with discrete particles.
The disadvantages are:
- Requires extensive process validation testing.
- Complex control system.


Return to: Page Top | Inspection Start