

High Purity Water System (7/93)
GUIDE TO INSPECTIONS OF HIGH PURITY WATER SYSTEMS
Note: This document is reference material for investigators and other FDA personnel. The document does not bind FDA, and does no confer any rights, privileges, benefits, or immunities for or on any person(s).
This guide discusses, primarily from a microbiological aspect, the review and evaluation of high purity water systems that are used for the manufacture of drug products and drug substances. It also includes a review of the design of the various types of systems and some of the problems that have been associated with these systems. As with other guides, it is not all-inclusive, but provides background and guidance for the review and evaluation of high purity water systems. The Guide To Inspections of Microbiological Pharmaceutical Quality Control Laboratories (May, 1993) provides additional guidance.
I. SYSTEM DESIGN
One of the basic considerations in the design of a system is the type of product that is to be manufactured. For parenteral products where there is a concern for pyrogens, it is expected that Water for Injection will be used. This applies to the formulation of products, as well as to the final washing of components and equipment used in their manufacture. Distillation and Reverse Osmosis (RO) filtration are the only acceptable methods listed in the USP for producing Water for Injection. However, in the bulk Pharmaceutical and Biotechnology industries and some foreign companies, Ultra Filtration (UF) is employed to minimize endotoxins in those drug substances that are administered parenterally.
For some ophthalmic products, such as the ophthalmic irrigating solution, and some inhalation products, such as Sterile Water for Inhalation, where there are pyrogen specifications, it is expected that Water for Injection be used in their formulation. However, for most inhalation and ophthalmic products, purified water is used in their formulation. This also applies to topicals, cosmetics and oral products.
Another design consideration is the temperature of the system. It is recognized that hot (65 - 80oC) systems are self sanitizing. While the cost of other systems may be less expensive for a company, the cost of maintenance, testing and potential problems may be greater than the cost of energy saved. Whether a system is circulating or one-way is also an important design consideration. Obviously, water in constant motion is less liable to have high levels of contaminant. A one-way water system is basically a "dead-leg".
Finally, and possibly the most important consideration, is the risk assessment or level of quality that is desired. It should be recognized that different products require different quality waters. Parenterals require very pure water with no endotoxins. Topical and oral products require less pure water and do not have a requirement for endotoxins. Even with topical and oral products there are factors that dictate different qualities for water. For example, preservatives in antacids are marginally effective, so more stringent microbial limits have to be set. The quality control department should assess each product manufactured with the water from their system and determine the microbial action limits based on the most microbial sensitive product. In lieu of stringent water action limits in the system the manufacturer can add a microbial reduction step in the manufacturing process for the sensitive drug product(s).
II. SYSTEM VALIDATION
A basic reference used for the validation of high purity water systems is the Parenteral Drug Association Technical Report No. 4 titled, "Design Concepts for the Validation of a Water for Injection System."
The introduction provides guidance and states that, "Validation often involves the use of an appropriate challenge. In this situation, it would be undesirable to introduce microorganisms into an on-line system; therefore, reliance is placed on periodic testing for microbiological quality and on the installation of monitoring equipment at specific checkpoints to ensure that the total system is operating properly and continuously fulfilling its intended function."
In the review of a validation report, or in the validation of a high purity water system, there are several aspects that should be considered. Documentation should include a description of the system along with a print. The drawing needs to show all equipment in the system from the water feed to points of use. It should also show all sampling points and their designations. If a system has no print, it is usually considered an objectionable condition. The thinking is if there is no print, then how can the system be validated? How can a quality control manager or microbiologist know where to sample? In those facilities observed without updated prints, serious problems were identified in these systems. The print should be compared to the actual system annually to insure its accuracy, to detect unreported changes and confirm reported changes to the system.
After all the equipment and piping has been verified as installed correctly and working as specified, the initial phase of the water system validation can begin. During this phase the operational parameters and the cleaning/ sanitization procedures and frequencies will be developed. Sampling should be daily after each step in the purification process and at each point of use for two to four weeks. The sampling procedure for point of use sampling should reflect how the water is to be drawn e.g. if a hose is usually attached the sample should be taken at the end of the hose. If the SOP calls for the line to be flushed before use of the water from that point, then the sample is taken after the flush. At the end of the two to four week time period the firm should have developed its SOPs for operation of the water system.
The second phase of the system validation is to demonstrate that the system will consistently produce the desired water quality when operated in conformance with the SOPs. The sampling is performed as in the initial phase and for the same time period. At the end of this phase the data should demonstrate that the system will consistently produce the desired quality of water.
The third phase of validation is designed to demonstrate that when the water system is operated in accordance with the SOPs over a long period of time it will consistently produce water of the desired quality. Any variations in the quality of the feedwater that could affect the operation and ultimately the water quality will be picked up during this phase of the validation. Sampling is performed according to routine procedures and frequencies. For Water for Injection systems the samples should be taken daily from a minimum of one point of use, with all points of use tested weekly. The validation of the water system is completed when the firm has a full years worth of data.
While the above validation scheme is not the only way a system can be validated, it contains the necessary elements for validation of a water system. First, there must be data to support the SOPs. Second, there must be data demonstrating that the SOPs are valid and that the system is capable of consistently producing water that meets the desired specifications. Finally, there must be data to demonstrate that seasonal variations in the feedwater do not adversely affect the operation of the system or the water quality.
The last part of the validation is the compilation of the data, with any conclusions into the final report. The final validation report must be signed by the appropriate people responsible for operation and quality assurance of the water system.
A typical problem that occurs is the failure of operating procedures to preclude contamination of the system with non-sterile air remaining in a pipe after drainage. In a system illustrated as in Figure 1, (below) a typical problem occurs when a washer or hose connection is flushed and then drained at the end of the operation. After draining, this valve (the second off of the system) is closed. If on the next day or start-up of the operation the primary valve off of the circulating system is opened, then the non-sterile air remaining in the pipe after drainage would contaminate the system. The solution is to pro-vide for operational procedures that provide for opening the secondary valve before the primary valve to flush the pipe prior to use.

Another major consideration in the validation of high purity water systems is the acceptance criteria. Consistent results throughout the system over a period of time constitute the primary element.
III. MICROBIAL LIMITS
Water For Injection Systems
Regarding microbiological results, for Water For Injection, it is expected that they be essentially sterile. Since sampling frequently is performed in non-sterile areas and is not truly aseptic, occasional low level counts due to sampling errors may occur. Agency policy, is that less than 10 CFU/100ml is an acceptable action limit. None of the limits for water are pass/fail limits. All limits are action limits. When action limits are exceeded the firm must investigate the cause of the problem, take action to correct the problem and assess the impact of the microbial contamination on products manufactured with the water and document the results of their investigation.
With regard to sample size, 100 - 300 mL is preferred when sampling Water for Injection systems. Sample volumes less than 100 mL are unacceptable.
The real concern in WFI is endotoxins. Because WFI can pass the LAL endotoxin test and still fail the above microbial action limit, it is important to monitor WFI systems for both endotoxins and microorganisms.
Purified Water Systems
For purified water systems, microbiological specifications are not as clear. USP XXII specifications, that it complies with federal Environmental Protection Agency regulations for drinking water, are recognized as being minimal specifications. There have been attempts by some to establish meaningful microbiological specifications for purified water. The CFTA proposed a specification of not more than 500 organisms per ml. The USP XXII has an action guideline of not greater than 100 organisms per ml. Although microbiological specifications have been discussed, none (other than EPA standards) have been established. Agency policy is that any action limit over 100 CFU/mL for a purified water system is unacceptable.
The purpose of establishing any action limit or level is to assure that the water system is under control. Any action limit established will depend upon the overall purified water system and further processing of the finished product and its use. For example, purified water used to manufacture drug products by cold processing should be free of objectionable organisms. We have defined "objectionable organisms" as any organisms that can cause infections when the drug product is used as directed or any organism capable of growth in the drug product. As pointed out in the Guide to Inspections of Microbiological Pharmaceutical Quality Control Laboratories, the specific contaminant, rather than the number is generally more significant.
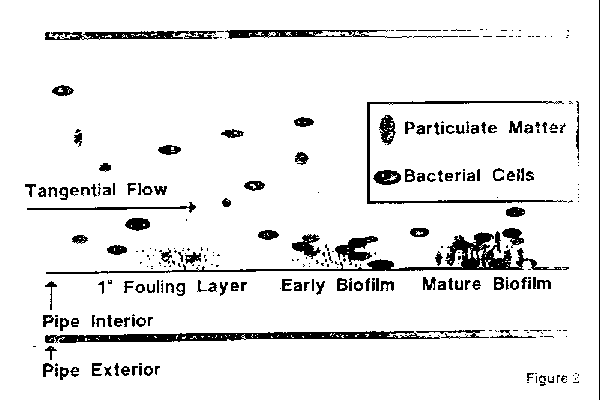
Organisms exist in a water system either as free floating in the water or attached to the walls of the pipes and tanks. When they are attached to the walls they are known as biofilm, which continuously slough off organisms. Thus, contamination is not uniformly distributed in a system and the sample may not be representative of the type and level of contamination. A count of 10 CFU/mL in one sample and 100 or even 1000 CFU/mL in a subsequent sample would not be unrealistic.
Thus, in establishing the level of contamination allowed in a high purity water system used in the manufacture of a non-sterile product requires an understanding of the use of the product, the formulation (preservative system) and manufacturing process. For example, antacids, which do not have an effective preservative system, require an action limit below the 100 CFU/mL maximum.
The USP gives some guidance in their monograph on Microbiological Attributes of Non-Sterile Products. It points out that, "The significance of microorganisms in non-sterile pharmaceutical products should be evaluated in terms of the use of the product, the nature of the product, and the potential harm to the user." Thus, not just the indicator organisms listed in some of the specific monographs present problems. It is up to each manufacturer to evaluate their product, the way it is manufactured, and establish am acceptable action level of contamination, not to exceed the maximum, for the water system, based on the highest risk product manufactured with the water.
IV. WATER FOR INJECTION SYSTEMS
In the review and evaluation of Water For Injection systems, there are several concerns.
Pretreatment of feedwater is recommended by most manufacturers of distillation equipment and is definitely required for RO units. The incoming feedwater quality may fluctuate during the life of the system depending upon seasonal variations and other external factors beyond the control of the pharmaceutical facility. For example, in the spring (at least in the N.E.), increases in gram negative organisms have been known. Also, new construction or fires can cause a depletion of water stores in old mains which can cause an influx of heavily contaminated water of a different flora.
A water system should be designed to operate within these anticipated extremes. Obviously, the only way to know the extremes is to periodically monitor feedwater. If the feedwater is from a municipal water system, reports from the municipality testing can be used in lieu of in-house testing.
V. STILL
Figures 3-5 represent a typical basic diagram of a WFI system. Most of the new systems now use multi-effect stills. In some of the facilities, there has been evidence of endotoxin contamination. In one system this occurred, due to malfunction of the feedwater valve and level control in the still which resulted in droplets of feedwater being carried over in the distillate.
In another system with endotoxin problems, it was noted that there was approximately 50 liters of WFI in the condenser at the start-up. Since this water could lie in the condenser for up to several days (i.e., over the weekend), it was believed that this was the reason for unacceptable levels of endotoxins.
More common, however, is the failure to adequately treat feedwater to reduce levels of endotoxins. Many of the still fabricators will only guarantee a 2.5 log to 3 log reduction in the endotoxin content. Therefore, it is not surprising that in systems where the feedwater occasionally spikes to 250 EU/ml, unacceptable levels of endotoxins may occasionally appear in the distillate (WFI). For example, recently three new stills, including two multi-effect, were found to be periodically yielding WFI with levels greater than .25 EU/ml. Pretreatment systems for the stills included only deionization systems with no UF, RO or distillation. Unless a firm has a satisfactory pretreatment system, it would be extremely difficult for them to demonstrate that the system is validated.
The above examples of problems with distillation units used to produce WFI, point to problems with maintenance of the equipment or improper operation of the system indicating that the system has not been properly validated or that the initial validation is no longer valid. If you see these types of problems you should look very closely at the system design, any changes that have been made to the system, the validation report and the routine test data to determine if the system is operating in a state of control.
Typically, conductivity meters are used on water systems to monitor chemical quality and have no meaning regarding microbiological quality.
Figures 3-5 also show petcocks or small sampling ports between each piece of equipment, such as after the still and before the holding tank. These are in the system to isolate major pieces of equipment. This is necessary for the qualification of the equipment and for the investigation of any problems which might occur.
VI. HEAT EXCHANGERS
One principal component of the still is the heat exchanger. Because of the similar ionic quality of distilled and deionized water, conductivity meters cannot be used to monitor microbiological quality. Positive pressure such as in vapor compression or double tubesheet design should be employed to prevent possible feedwater to distillate contamination in a leaky heat exchanger.
An FDA Inspectors Technical Guide with the subject of "Heat Exchangers to Avoid Contamination" discusses the design and potential problems associated with heat exchangers. The guide points out that there are two methods for preventing contamination by leakage. One is to provide gauges to constantly monitor pressure differentials to ensure that the higher pressure is always on the clean fluid side. The other is to utilize the double-tubesheet type of heat exchanger.
In some systems, heat exchangers are utilized to cool water at use points. For the most part, cooling water is not circulated through them when not in use. In a few situations, pinholes formed in the tubing after they were drained (on the cooling water side) and not in use. It was determined that a small amount of moisture remaining in the tubes when combined with air caused a corrosion of the stainless steel tubes on the cooling water side. Thus, it is recommended that when not in use, heat exchangers not be drained of the cooling water.
VII. HOLDING TANK
In hot systems, temperature is usually maintained by applying heat to a jacketed holding tank or by placing a heat exchanger in the line prior to an insulated holding tank.
The one component of the holding tank that generates the most discussion is the vent filter. It is expected that there be some program for integrity testing this filter to assure that it is intact. Typically, filters are now jacketed to prevent condensate or water from blocking the hydrophobic vent filter. If this occurs (the vent filter becomes blocked), possibly either the filter will rupture or the tank will collapse. There are methods for integrity testing of vent filters in place.
It is expected, therefore, that the vent filter be located in a position on the holding tank where it is readily accessible.
Just because a WFI system is relatively new and distillation is employed, it is not problem-free. In an inspection of a manufacturer of parenterals, a system fabricated in 1984 was observed. Refer to Figure 6. While the system may appear somewhat complex on the initial review, it was found to be relatively simple. Figure 7 is a schematic of the system. The observations at the conclusion of the inspection of this manufacturer included, "Operational procedures for the Water For Injection system failed to provide for periodic complete flushing or draining. The system was also open to the atmosphere and room environment. Compounding equipment consisted of non-sealed, open tanks with lids. The Water for Injection holding tank was also not sealed and was never sampled for endotoxins." Because of these and other comments, the firm recalled several products and discontinued operations.
VIII. PUMPS
Pumps burn out and parts wear. Also, if pumps are static and not continuously in operation, their reservoir can be a static area where water will lie. For example, in an inspection, it was noted that a firm had to install a drain from the low point in a pump housing. Pseudomonas sp. contamination was periodically found in their water system which was attributed in part to a pump which only periodically is operational.
IX. PIPING
Piping in WFI systems usually consist of a high polished stainless steel. In a few cases, manufacturers have begun to utilize PVDF (polyvinylidene fluoride) piping. It is purported that this piping can tolerate heat with no extractables being leached. A major problem with PVDF tubing is that it requires considerable support. When this tubing is heated, it tends to sag and may stress the weld (fusion) connection and result in leakage. Additionally, initially at least, fluoride levels are high. This piping is of benefit in product delivery systems where low level metal contamination may accelerate the degradation of drug product, such as in the Biotech industry.
One common problem with piping is that of "dead-legs". The proposed LVP Regulations defined dead-legs as not having an unused portion greater in length than six diameters of the unused pipe measured from the axis of the pipe in use. It should be pointed out that this was developed for hot 75 - 80o circulating systems. With colder systems (65 - 75oC), any drops or unused portion of any length of piping has the potential for the formation of a biofilm and should be eliminated if possible or have special sanitizing procedures. There should be n o threaded fittings in a pharmaceutical water system. All pipe joints must utilize sanitary fittings or be butt welded. Sanitary fittings will usually be used where the piping meets valves, tanks and other equipment that must be removed for maintenance or replacement. Therefore, the firm's procedures for sanitization, as well as the actual piping, should be reviewed and evaluated during the inspection.
X. REVERSE OSMOSIS
Another acceptable method for manufacturing Water for Injection is Reverse Osmosis (RO). However, because these systems are cold, and because RO filters are not absolute, microbiological contamination is not unusual. Figure 8 shows a system that was in use several years ago. There are five RO units in this system which are in parallel. Since RO filters are not absolute, the filter manufacturers recommend that at least two be in series. The drawing also illustrates an Ultraviolet (UV) light in the system downstream from the RO units. The light was needed to control microbiological contamination.
Also in this system were ball valves. These valves are not considered sanitary valves since the center of the valve can have water in it when the valve is closed. This is a stagnant pool of water that can harbor microorganisms and provide a starting point for a biofilm.
As an additional comment on RO systems, with the recognition of microbiological problems, some manufacturers have installed heat exchangers immediately after the RO filters to heat the water to 75 - 80oC to minimize microbiological contamination.
With the development of biotechnology products, many small companies are utilizing RO and UF systems to produce high purity water. For example, Figure 9 illustrates a wall mounted system that is fed by a single pass RO unit.
As illustrated, most of these systems employ PVC or some type of plastic tubing. Because the systems are typically cold, the many joints in the system are subject to contamination. Another potential problem with PVC tubing is extractables. Looking at the WFI from a system to assure that it meets USP requirements without some assurance that there are no extractables would not be acceptable.
The systems also contain 0.2 micron point of use filters which can mask the level of microbiological contamination in the system. While it is recognized that endotoxins are the primary concern in such a system, a filter will reduce microbiological contamination, but not necessarily endotoxin contamination. If filters are used in a water system there should be a stated purpose for the filter, i.e., particulate removal or microbial reduction, and an SOP stating the frequency with which the filter is to be changed which is based on data generated during the validation of the system.
As previously discussed, because of the volume of water actually tested (.1ml for endotoxins vs. 100ml for WFI), the microbiological test offers a good index of the level of contamination in a system. Therefore, unless the water is sampled prior to the final 0.2 micron filter, microbiological testing will have little meaning.
At a reinspection of this facility, it was noted that they corrected the deficient water system with a circulating stainless steel piping system that was fed by four RO units in series. Because this manufacturer did not have a need for a large amount of water (the total system capacity was about 30 gallons), they attempted to let the system sit for approximately one day. Figure 9 shows that at zero time (at 9 AM on 3/10), there were no detectable levels of microorganisms and of endotoxins. After one day, this static non-circulating system was found to be contaminated. The four consecutive one hour samples also illustrate the variability among samples taken from a system. After the last sample at 12 PM was collected, the system was resanitized with 0.5% peroxide solution, flushed, recirculated and resampled. No levels of microbiological contamination were found on daily samples after the system was put back in operation. This is the reason the agency has recommended that non-recirculating water systems be drained daily and water not be allowed to sit in the system.
XI. PURIFIED WATER SYSTEMS
Many of the comments regarding equipment for WFI systems are applicable to Purified Water Systems. One type system that has been used to control microbiological contamination utilizes ozone. Figure 10 illustrates a typical system. Although the system has purported to be relatively inexpensive, there are some problems associated with it. For optimum effectiveness, it is required that dissolved ozone residual remain in the system. This presents both employee safety problems and use problems when drugs are formulated.
Published data for Vicks Greensboro, NC facility showed that their system was recontaminated in two to three days after the ozone generator was turned off. In an inspection of another manufacturer, it was noted that a firm was experiencing a contamination problem with Pseudomonas sp. Because of potential problems with employee safety, ozone was removed from the water prior to placing it in their recirculating system. It has been reported that dissolved ozone at a level of 0.45 mg/liter will remain in a system for a maximum of five to six hours.
Another manufacturer, as part of their daily sanitization, removes all drops off of their ozonated water system and disinfects them in filter sterilized 70% isopropyl alcohol. This manufacturer has reported excellent microbiological results. However, sampling is only performed immediately after sanitization and not at the end of operations. Thus, the results are not that meaningful.
Figure 11 and Figure12 illustrate another purified water system which had some problems. Unlike most of the other systems discussed, this is a one-way and not recirculating system. A heat exchanger is used to heat the water on a weekly basis and sanitize the system. Actually, the entire system is a "dead-leg."
Figure 11 also shows a 0.2 micron in line filter used to sanitize the purified water on a daily basis. In addition to the filter housing providing a good environment for microbiological contamination, a typical problem is water hammer that can cause "ballooning" of the filter. If a valve downstream from the filter is shut too fast, the water pressure will reverse and can cause "ballooning". Pipe vibration is a typical visible sign of high back pressure while passage of upstream contaminants on the filter face is a real problem. This system also contains several vertical drops at use points. During sanitization, it is important to "crack" the terminal valves so that all of the elbows and bends in the piping are full of water and thus, get complete exposure to the sanitizing agent.
It should be pointed out that simply because this is a one-way system, it is not inadequate. With good Standard Operational Procedures, based on validation data, and routine hot flushings of this system, it could be acceptable. A very long system (over 200 yards) with over 50 outlets was found acceptable. This system employed a daily flushing of all outlets with 80oC water.
The last system to be discussed is a system that was found to be objectionable. Pseudomonas sp. found as a contaminant in the system (after FDA testing) was also found in a topical steroid product (after FDA testing). Product recall and issuance of a Warning Letter resulted. This system (Figure 13) is also one-way that employs a UV light to control microbiological contamination. The light is turned on only when water is needed. Thus, there are times when water is allowed to remain in the system. This system also contains a flexible hose which is very difficult to sanitize. UV lights must be properly maintained to work. The glass sleeves around the bulb(s) must be kept clean or their effectiveness will decrease. In multibulb units there must be a system to determine that each bulb is functioning. It must be remembered that at best UV light will only kill 90% of the organisms entering the unit.
XIII. PROCESS WATER
Currently, the USP, pg. 4, in the General Notices Section, allows drug substances to be manufactured from Potable Water. It comments that any dosage form must be manufactured from Purified Water, Water For Injection, or one of the forms of Sterile Water. There is some inconsistency in these two statements, since Purified Water has to be used for the granulation of tablets, yet Potable Water can be used for the final purification of the drug substance.
The FDA Guide to Inspection of Bulk Pharmaceutical Chemicals comments on the concern for the quality of the water used for the manufacture of drug substances, particularly those drug substances used in parenteral manufacture. Excessive levels of microbiological and/or endotoxin contamination have been found in drug substances, with the source of contamination being the water used in purification. At this time, Water For Injection does not have to be used in the finishing steps of synthesis/purification of drug substances for parenteral use. However, such water systems used in the final stages of processing of drug substances for parenteral use should be validated to assure minimal endotoxin/ microbiological contamination.
In the bulk drug substance industry, particularly for parenteral grade substances, it is common to see Ultrafiltration (UF) and Reverse Osmosis (RO) systems in use in water systems. While ultrafiltration may not be as efficient at reducing pyrogens, they will reduce the high molecular weight endotoxins that are a contaminant in water systems. As with RO, UF is not absolute, but it will reduce numbers. Additionally, as previously discussed with other cold systems, there is considerable maintenance required to maintain the system.
For the manufacture of drug substances that are not for parenteral use, there is still a microbiological concern, although not to the degree as for parenteral grade drug substances. In some areas of the world, Potable (chlorinated) water may not present a microbiological problem. However, there may be other issues. For example, chlorinated water will generally increase chloride levels. In some areas, process water may be obtained directly from neutral sources.
In one inspection, a manufacturer was obtaining process water from a river located in a farming region. At one point, they had a problem with high levels of pesticides which was a run-off from farms in the areas. The manufacturing process and analytical methodology was not designed to remove and identify trace pesticide contaminants. Therefore, it would seem that this process water when used in the purification of drug substances would be unacceptable.
XIV. INSPECTION STRATEGY
Manufacturers typically will have periodic printouts or tabulations of results for their purified water systems. These printouts or data summaries should be reviewed. Additionally, investigation reports, when values exceed limits, should be reviewed.
Since microbiological test results from a water system are not usually obtained until after the drug product is manufactured, results exceeding limits should be reviewed with regard to the drug product formulated from such water. Consideration with regard to the further processing or release of such a product will be dependent upon the specific contaminant, the process and the end use of the product. Such situations are usually evaluated on a case-by-case basis. It is a good practice for such situations to include an investigation report with the logic for release/rejection discussed in the firm's report. End product microbiological testing, while providing some information should not be relied upon as the sole justification for the release of the drug product. The limitations of microbiological sampling and testing should be recognized.
Manufacturers should also have maintenance records or logs for equipment, such as the still. These logs should also be reviewed so that problems with the system and equipment can be evaluated.
In addition to reviewing test results, summary data, investigation reports and other data, the print of the system should be reviewed when conducting the actual physical inspection. As pointed out, an accurate description and print of the system is needed in order to demonstrate that the system is validated.
Return to: Page Top | Inspection Start